羧酸衍生物
费舍尔在酸性条件下酯化-羧酸酯
最后更新:2023年5月22日|
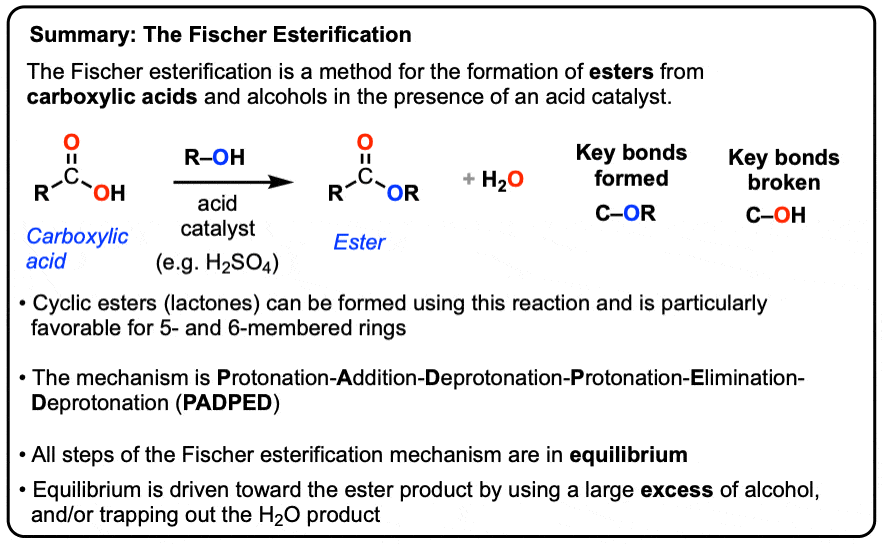
- 的转换羧酸到一个酯在酸性条件下通常被称为费歇尔酯化
- 在这个反应有一个平衡起始原料(羧酸+酒精)和产品(酯+水)。
- 右边的平衡驱动(酯通过使用大量过剩)酒精并通过移除任何水形成通过使用干燥剂(防潮珠)或通过切除Dean-Stark类型装置
- 循环酯类可以在这些条件下形成的,这被称为内酯。
- 这个反应的机制Protonation -一个ddition -Deprotonation -Protonation -Elimination -Deprotonation (PADPED)。
- 可以运行在相反方向的反应治疗酯多余的水的酸的存在。这是酸性的酯水解。
表的内容
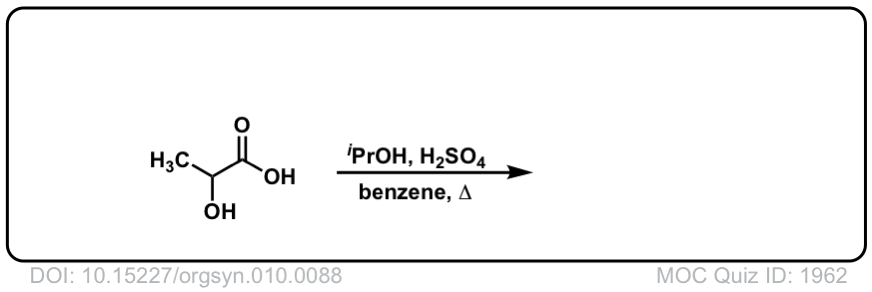
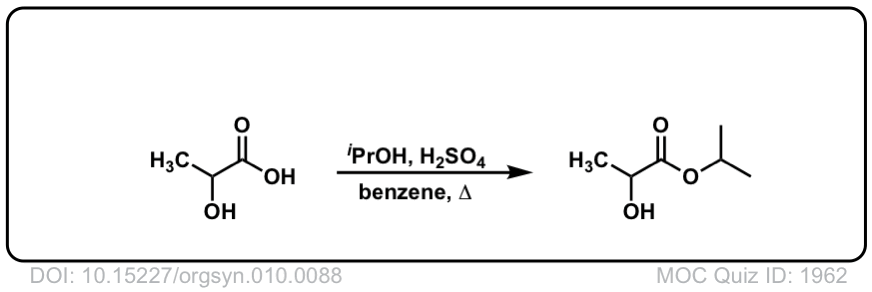
1。转换的羧酸酯(费舍尔酯化)
这就是所谓的费舍尔酯化。或者只是“酸酯化”。
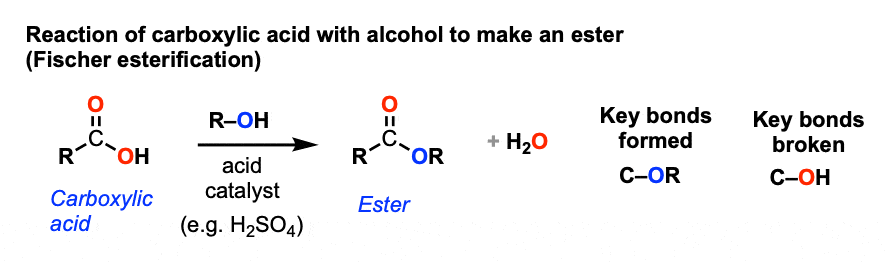
常见的酸的选择包括硫酸(H2所以4),除了酸(TsOH)和盐酸盐酸等等。对我们来说这些条件可以被认为是等价的。
这里有一些例子的费舍尔酯化。
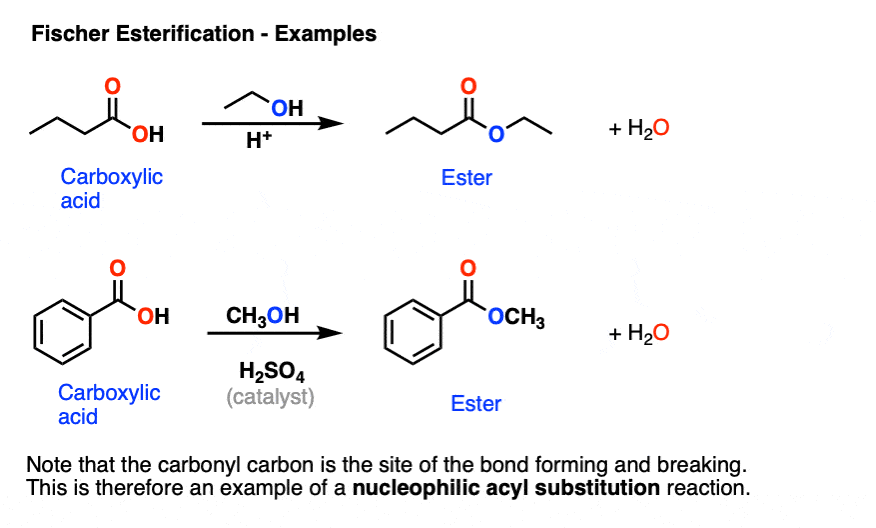
我们注意到在这个反应形式一个新的债券,我们切断打破一个切断键。
重要的是,我们正在形成,打破债券从氧气羰基,不债券从氧到R组。
因此,费舍尔酯化的一个例子亲核酰取代。
我们也形成两个新的债券地生成一个水分子。
2。分子内费舍尔酯化
费歇尔酯化反应也可以用来制造循环酯类(内酯)。
这是一个一个的例子分子内的的反应。的债券形式和破坏是完全一样的!但由于亲核试剂和亲电试剂连接到相同的分子,我们获得一个吗循环产品。
环的形成最适合5 -和六元环的形成。(注1)3和4元环通常太紧张形式通过这种方法,尽管还有其他方法4-membered的形成内酯环(注2]。
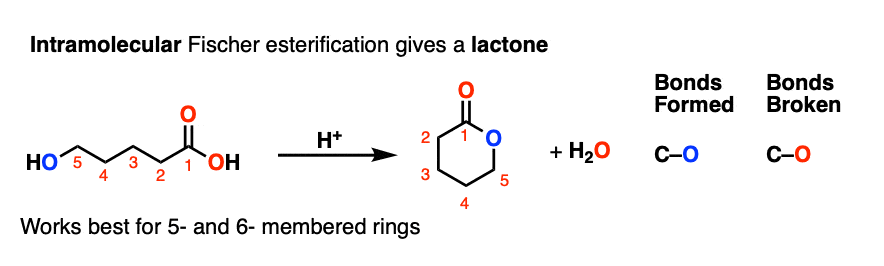
注意,在这个反应一个等价的水形成。
3所示。费歇尔酯化反应的机制
所以这个反应是如何工作的呢?
费歇尔酯化机理有6个步骤。每一步都是可逆的,起始原料和最终产品都处于平衡状态。
在费舍尔的第一步酯化,羰基氧气与酸质子化了的给一个氧鎓离子。
由此产生的质子化了的羰基是一个更好的亲电试剂比一个中立的羰基碳。
第二步是除了中性的亲核试剂(卢)质子化了的羧酸(切断形式,打破切断(π))。这导致一个四面体中间。(看到帖子:亲核加成,羰基)
![]()
接下来的两步一起被称为“质子转移”因为他们导致的净运动H +氧气从一个到另一个地方。去质子化的地酒精紧随其后的是质子化作用地的氧气。
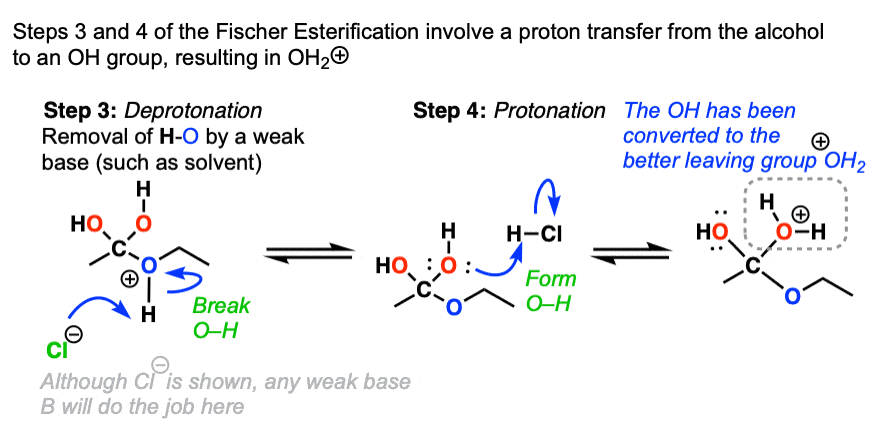
这导致形成一个好的离去基团(H2O)。消除H2O (形式切断(π),打破切断)给出了质子化了的酯。去质子化的酯然后给我们中性的酯产品(和水)。
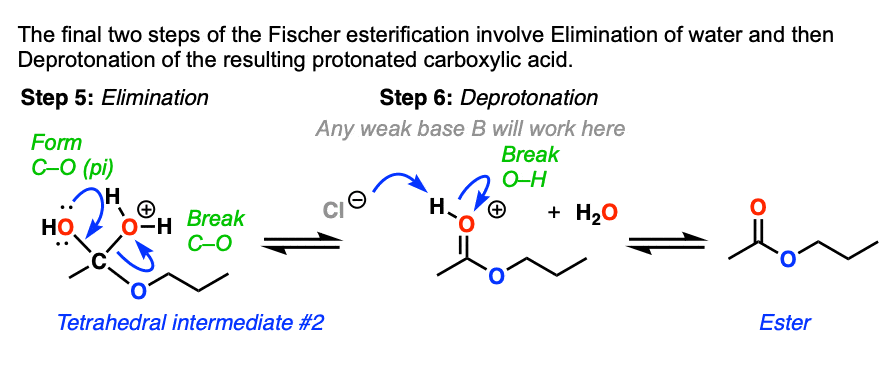
一个有用的完整机制是缩写PADPED助记符。
(Protonation-addition-deprotonation-protonation-elimination-deprotonation)。
这也是催化酯交换的机制(看到帖子:Tranesterification),以及酸性酯水解、形成亚胺和其他几个反应(看到帖子:学习6的价格机制)
4所示。比较费歇尔酯化和皂化
为什么发生酯化反应生成的酸在酸性条件下工作得很好,但是失败在基本条件下?
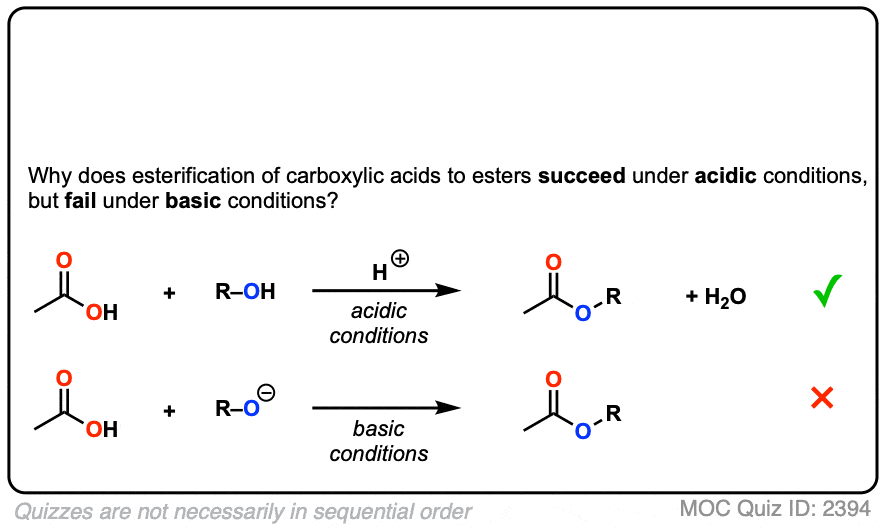 点击翻转
点击翻转
回想一下,一个主要推动力量对亲核酰取代反应强烈有利的当亲核试剂是一个强大的基础比吗离去基团。(看到帖子:亲核酰取代]
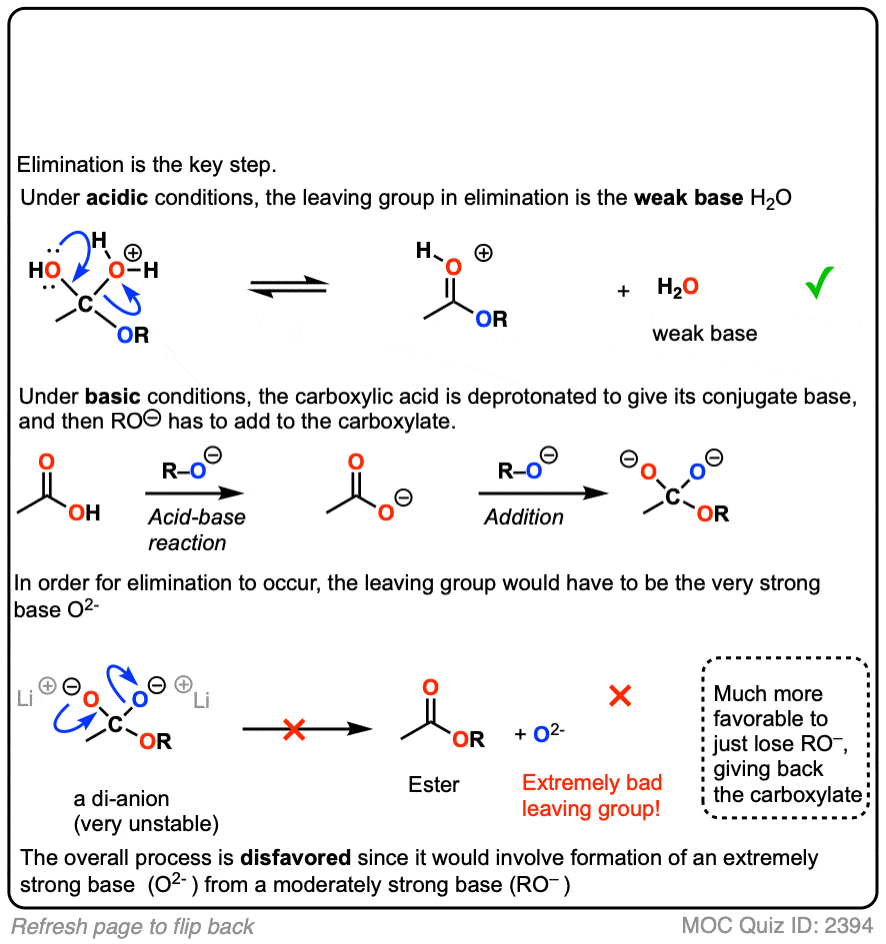
当我们使用酸性我们的条件亲核试剂卢武铉和我们离去基团是H2o .没有很强的动力的一种方式,但至少它不是不利的。
在基本条件下消除的关键一步是极其困难的。
如果RO(-)导致的位移(-),这将是一个好亲核酰替换。但由于羧酸是酸,酸碱反应之间的羧酸和RO(-)发生第一次,给了羧酸盐有数2(-)。
为了让亲核酰替换上发生羧酸盐,离去基团必须是O (2 -)。太不利了,因为这是一个更强大的比RO(-)基地!所以让这个反应工作希望向后流动的河。
所以使用酸这个反应让一切变得不同了。自共轭酸是一个更好的离去基团(看到帖子:是什么让一个好的离去基团)使用一个酸催化剂使我们能够执行亲核酰取代基的情况下就会失败。
5。的驱动力是什么?
还有最后一个问题值得一问。有什么实际的费舍尔酯化的动力吗?
通常,亲核酰取代反应遵循酸碱平庸的原则。这意味着反应所得的方向较强的基础(亲核试剂)取代较弱的基础(离去基团)。(看到帖子:亲核酰取代]
但在这里,离开组织前进的方向(H2O)和反向(卢武铉)大致相同离去基团能力。
为什么应该遵循任何方向?
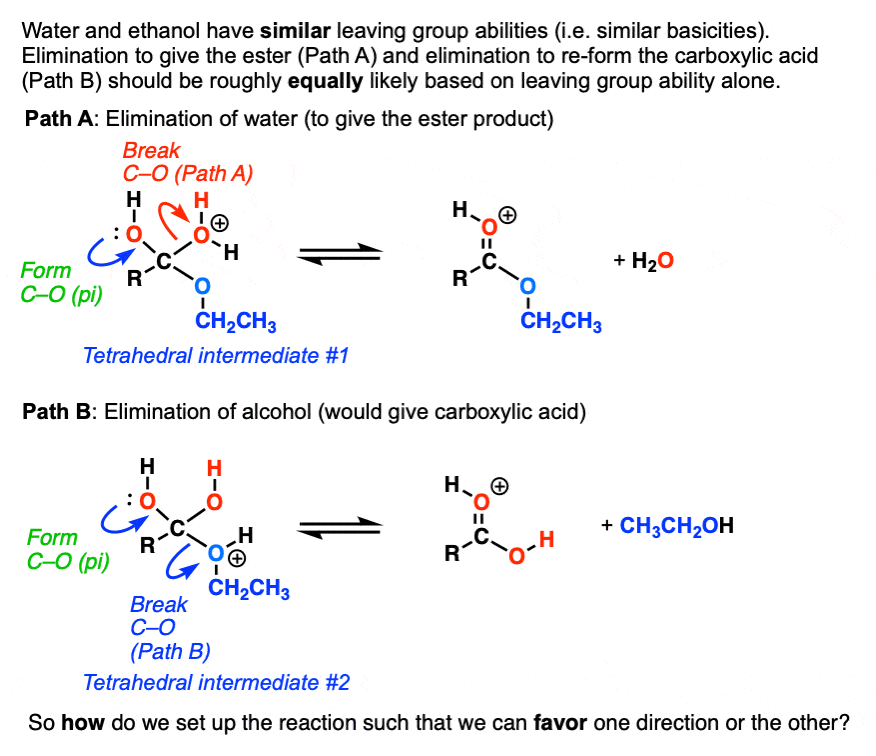
好问题!由于没有天生的强大动力反应,我们将不得不选择我们的反应条件这种平衡会流的方向所需的产品。
主要有两种方法可以做到这一点,可以结合这两种方法。
6。大量多余的试剂
一项研究在这个反应注3)用乙酸和乙醇酸催化剂的存在。
发现运行费舍尔使用等量的乙酸和乙醇酯化给65%的收益率酯在平衡。
不坏。但反应可以进一步推动正确的使用超过10倍酒精。在这种情况下,他们获得了97%的收益酯。
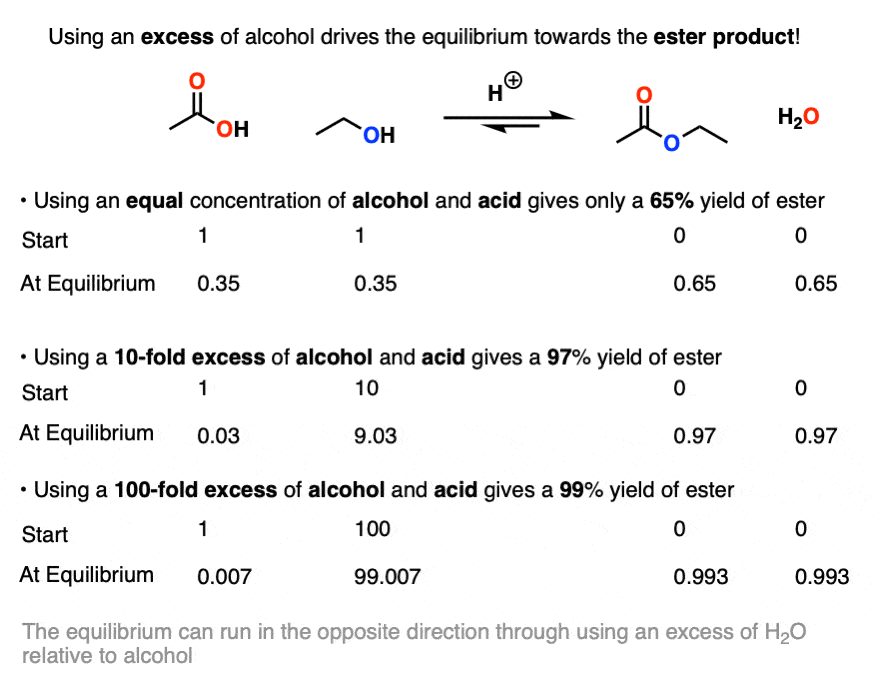
通过使用100倍超额,他们甚至能够驱动反应向99%的收益率。
7所示。去除水Dean-Stark陷阱
另一种方式,以确保反应的方向运行酯的形成是删除的水形成的。这推动平衡向正确的通过勒移动的原理。
一个聪明的方法是使用一个名为Dean-Stark陷阱的装置。
在这个过程中,使用的是溶剂,如苯或甲苯。这些分子co-distill在一起,形成所谓的共沸混合物。蒸汽冷凝回流冷凝器的底部。此时,液体滴下来,水,比甲苯更密集,水槽底部的陷阱。
水从反应混合物中删除,逆反应(酯水解)呈现的是不可能的。
例如,看到:(免费)的这篇文章(Org Syn 1949, 29岁,33)
8。总结
费歇尔酯化是其中一个典型的反应在有机化学不会消失。
它是最便宜和最有效的的形成过程酯类从羧酸,特别是在大规模。
费歇尔酯化中的步骤都是潜在可逆的。六个步骤的机制(Protonation-Addition-Deprotonation-Deprotonation-Elimination-Deprotonation)也逆反应的六个步骤,催化水解为酯类。
笔记
注1。形成的平衡不牵强附会的,五年和六元环是非常有利的原因的。一个分子的离去基团(如H2O)释放到溶液和服务员平动熵的增加(3度的平移旋转熵的熵和三度)。这是有时被称为螯合效应。(裁判]
注2。Beta-lactones无法通过费舍尔酯化形成四元环有太多环应变。然而,beta-lactones可以通过分子内形成的关闭酒精在酸性氯化物或类似的羧酸衍生物与一个优秀的离去基团。Beta-lactones也发现一些天然产物。
注3。这张桌子是改编自499页的“有机化学概论”(第四版),编辑Streitweiser,和实验。(链接]
测试你自己!
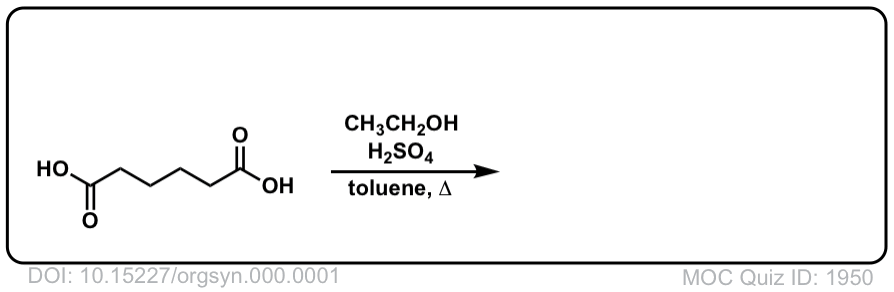 点击翻转
点击翻转
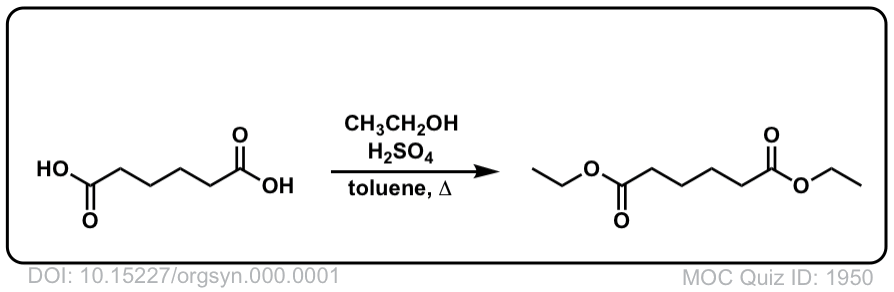
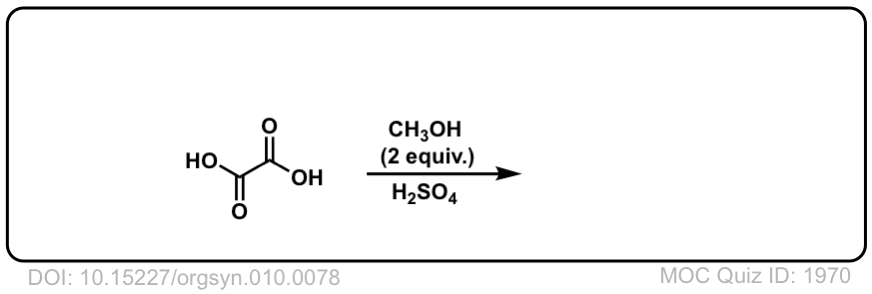
 点击翻转
点击翻转
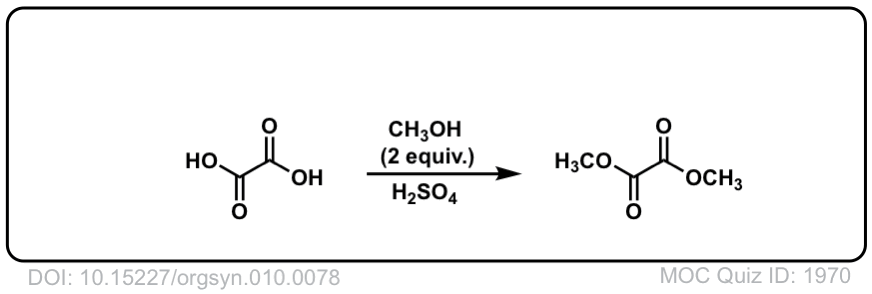
 点击翻转
点击翻转
(高级)引用和进一步阅读
引用
- 第一个例子
埃米尔•费舍尔亚瑟斯拜耳(1895年)。“Darstellung der酯”。
创刊号中。28日:3252 - 3258
DOI:1002 / cber.189502803176
原始论文由埃米尔费舍尔和亚瑟斯拜耳描述催化羧酸和醇的酯化反应。 - 质子的状态和地理酸催化酯化反应的机理
h·齐默尔曼博士博士j·鲁道夫
《应用化学》Int。问题1,卷4日,1965年1月,40至49页
DOI:10.1002 / anie.196500401
考虑质子迁移的浓缩阶段建议两步酯化反应的机理,并通过一个四面体中间收益。 - 己二酸乙酯
m . Micovic
Org。Synth。科尔。卷。2,264
DOI:10.15227 / orgsyn.000.0001
的第一个程序有机合成,一个可靠的来源可再生的有机转换。这个使用费舍尔将己二酸酯化,二酸和尼龙6的前兆,6、乙基己二酸。 - 亲核的反应的催化机制羧酸衍生品。
Myron l·本德
化学评论1960年60 (1),53 - 113
DOI:10.1021 / cr60203a005 - 苯基乙酸制备苯酚和乙酸:重新评估通用教科书的误解
m·b·霍金
《化学教育1980年57(7),527年
DOI:10.1021 / ed057p527 - 最近的事态发展在羧基酯化和保护的方法
埃德温·海斯蓝
四面体17、卷36问题,1980年,2409 - 2433
DOI:10.1016 / 0040 - 4020 (80)80219 - 5 - 熵的贡献在酶和分子内反应速度加速度和螯合效应
迈克尔·a·佩奇和威廉·p·Jencks
Proc, Nat。学会科学。1971年,68年(8),1678 - 1683
DOI:10.1073 / pnas.68.8.1678
螯合效应的讨论。当一个新的环形式,有一个旋转的限制引起的熵减少alongt碳碳键(约4.5熵单位/债券)但这是大幅增加足以弥补的平移自由度提供额外的分子的释放到解决方案。
评论
评论部分