有机金属化合物
格氏试剂的反应
最后更新:2023年2月28日|
格氏试剂的反应
- 格氏试剂是优秀的碳基的亲核试剂以及强大的基地。
- 他们将增加醛和酮醇形成质子化作用后(步骤)
- 他们将增加两次给叔醇酯。
- 他们将增加less-substituted环氧化合物
- 格氏试剂也将与二氧化碳反应(CO2)给羧酸(酸后检查)。
- 格氏试剂不会执行SN2与卤代烃反应。他们也不兼容羧酸或醇。
表的内容
1。提醒:格氏试剂的亲核试剂
到目前为止,在本系列中,我们已经介绍了有机金属化合物并说他们的碳亲核。我们学过如何让他们从烷基、烯基或芳基卤化物(以及某些方面不让他们!),发现它们很强的基地。
格氏是他们最有趣的地方是碳基的亲核试剂,因此我们可以把格氏试剂与各种亲电的碳物种形成新的碳碳键。
由于碳碳键构成的“骨干”分子在有机化学中,事实证明,这类反应是非常有用的。事实上,它赢得了它的发现者,维克多·格里纳1912年诺贝尔化学奖。
就我们的目的而言,关键碳基亲电试剂与格氏试剂反应环氧化合物、醛、酮、酯类。让我们去通过他们。
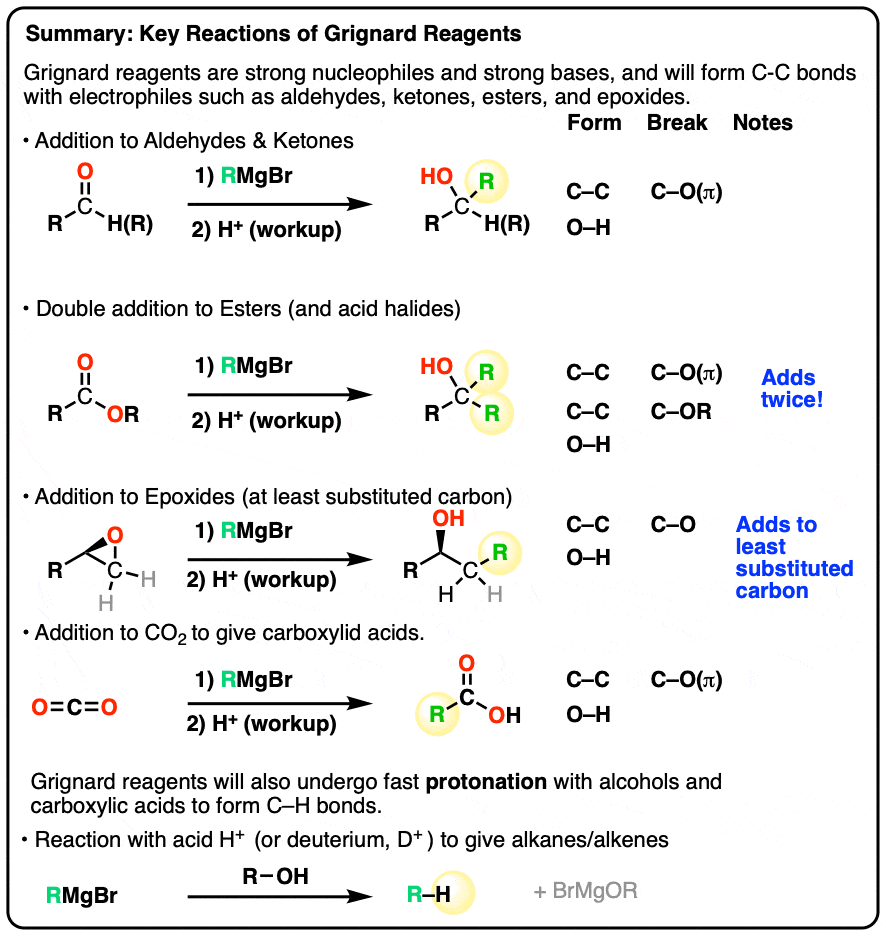
2。格氏试剂与环氧化合物
环氧化合物(“环氧乙烷”如果你是一个IUPAC stickler)是三人循环环醚具有相当大的压力。正如我们所看到的,这枚戒指压力使他们有点“弹簧”攻击的亲核试剂成立新债券,这将导致碳环的开放。
带负电的亲核试剂(如格氏)往往与环氧化合物反应的方式类似于年代N2反应:攻击发生在至少替换碳环氧化物。这里有一个例子:
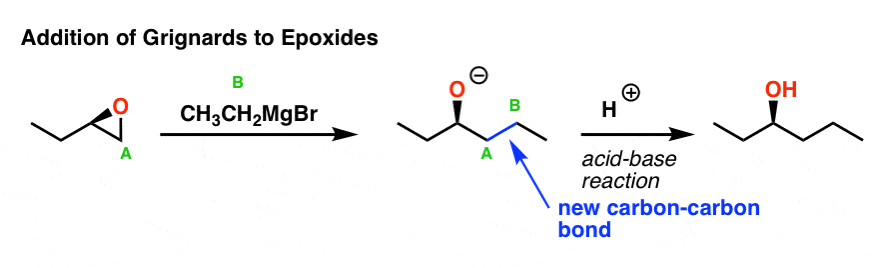
请注意这里形成的债券,打破了:我们形成一个新的碳碳键(a和B之间的碳),和打破了切断债券(碳和氧之间)。这导致了一个带负电荷的氧气(醇盐):生产最后的酒类产品,我们通常淬火反应与酸的来源,形成地。
这是反应是如何工作的。困难的是认识到的亲核试剂是一对电子C-Mg邦德:记得以前的帖子,碳强烈δ——(亲核),因为它更大的电负性比镁。
它可能是有用的作为CH想象下面的格氏试剂3CH2- - - - - -。除此之外,反应相当简单如果你看过一个年代N2反应之前:我们同时形成碳碳和切断。
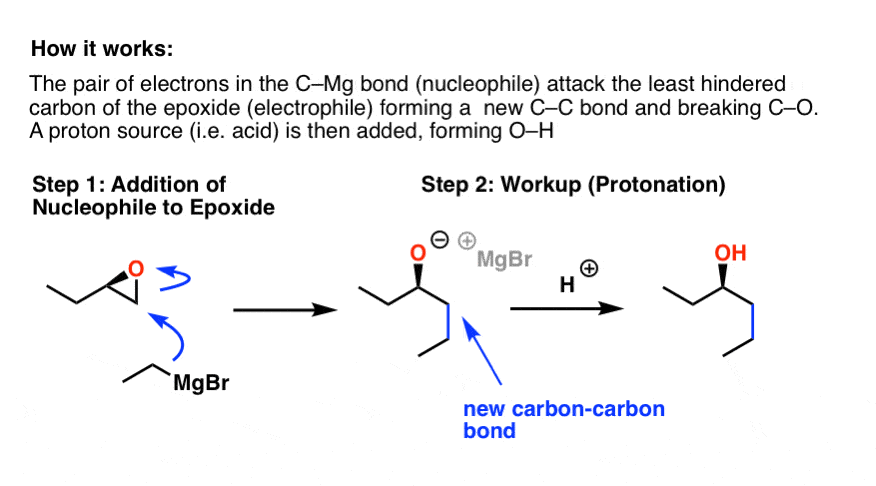
请注意,这个反应也形成了一个“醇盐”。为了获得我们的中性酒精产品结束时,我们必须执行第二步:“检查”(“淬火”)与酸的来源。这是写各种方式- H +, H3O + H2啊,还是“酸检查”。这一步发生后关键格氏反应,原因应该是显而易见的——强大的基地,格氏试剂是被酸。
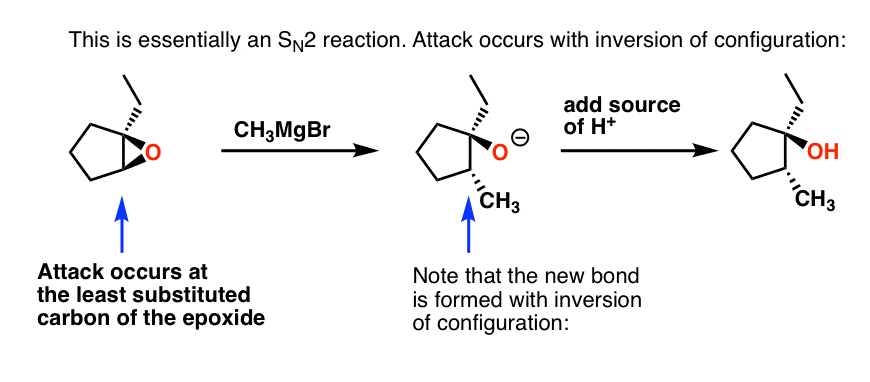
另一件事要记住立体化学的环氧.Consistent的年代N2的反应,如果反应发生在仲碳,我们将观察反演的配置:
3所示。格氏反应与醛和酮
第二课重要的亲电试剂与格氏反应(也可以说是最重要的类亲电试剂)醛和酮。如果你还没有覆盖这些官能团的反应,一个简短的摘要:羰基碳是一个亲电试剂,当在这个碳亲核试剂的反应,它伴随着乳沟的切断π键(π键)。(羰基加成机制上,看到帖子:亲核加成)
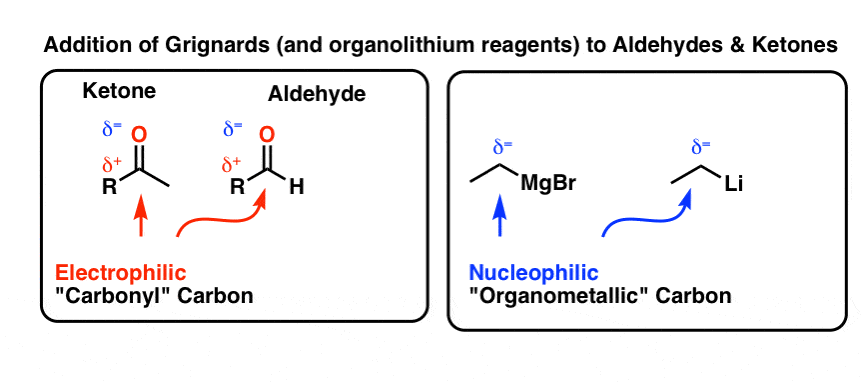
这里有一些例子与醛和酮的格氏反应。注意,在每种情况下我们正在形成一个新的债券之间的羰基碳(标签)和绑定到镁(称为B),我们打破了切断π键。
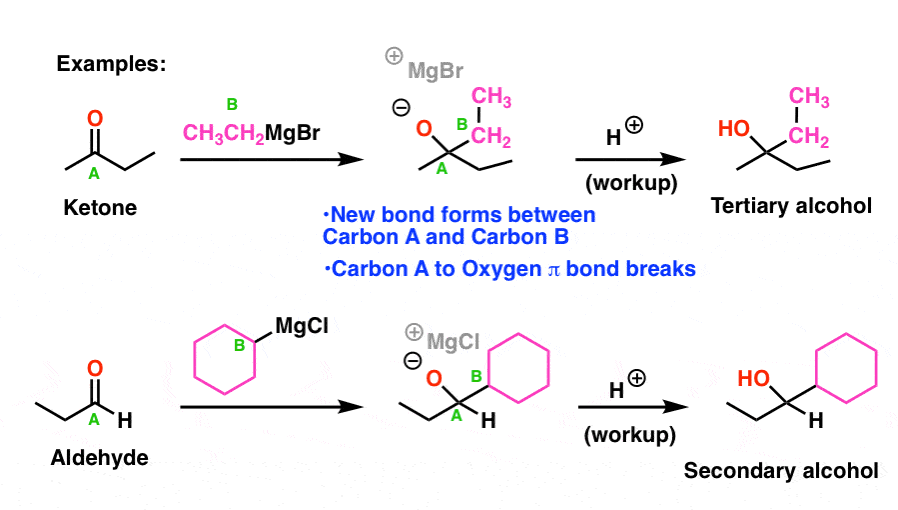
所以这个反应是如何工作的呢?
让我们熟悉一个非常重要的机制称为“除了”(有时称为“加法”)。这是到目前为止最重要的羰基的反应,如果你给自己一个鸡,每次你会看到变化的组织2,你会在你的房间里有很多鸡蛋的学期结束。
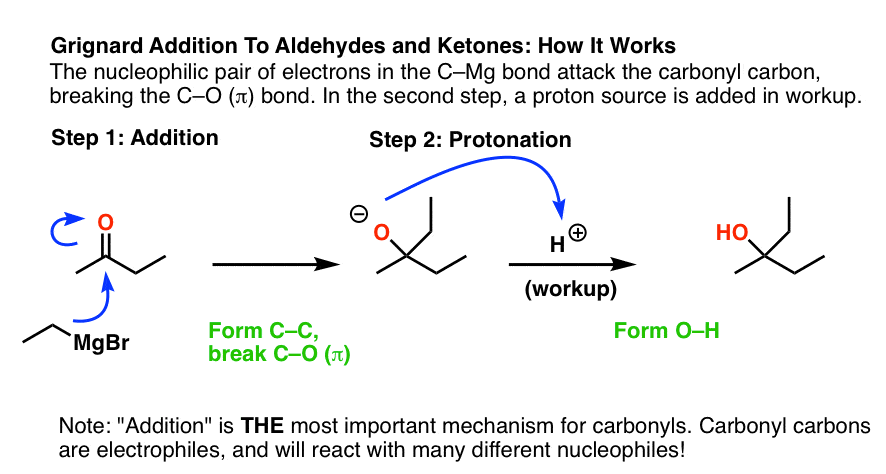
请注意,这个反应也形成了一个“醇盐”。为了获得我们的中性酒精产品结束时,我们必须执行一个“检查”(“淬火”)与酸的来源,形成地。
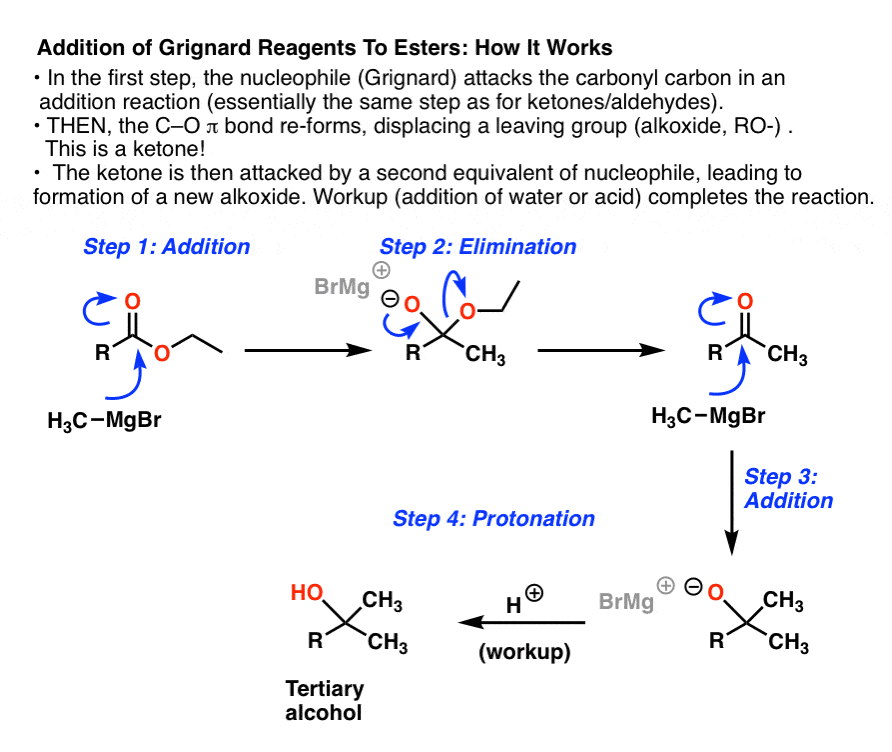
4所示。与酯类格氏试剂的反应
醛和酮的酯是近亲:他们由一个羰基直接连接到一个或一组。如您所料,他们在类似的方式与格氏反应醛和酮:形成新的碳碳键和破损的切断(π键)。
然而,有转折的反应酯与醛和酮不存在。仔细观察:有什么不同吗?
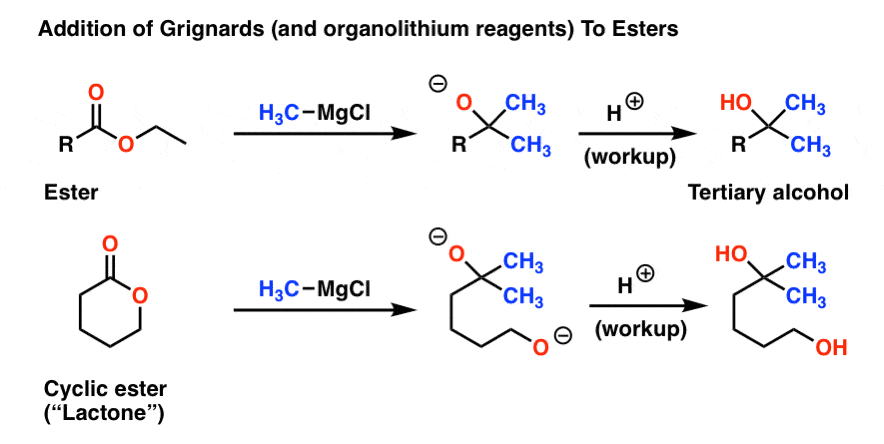
注意,在这两种情况下我们补充道两个酯等价物的格氏试剂,形成三级醇。
等一下——这是怎么发生的? !
5。为什么会有“双加法”与酯类?的机制
这个反应了第二个最重要的机理,羰基(“加法”旁边),即“消除”。事实上“消灭”是完全相反的“加法”!让我们穿过它。有4个步骤
- 在第一步格氏执行除了酯反应,形成碳碳和打破切断(π),给了我们一个中间的带负电荷的氧气。我们已经见过这种类型的反应增加格氏醛和酮。
- 现在是新的步骤:消除消除(有时,“1、2”)。这中间有一个相当不错离去基团(哟2CH3在下面的情况下)。接下来会发生什么是切断π键的改革与离去基团的驱逐(CH3CH2O- - - - - -在下面的情况下)。换句话说,我们切断形成π和打破切断单键。新产品是一个酮。
在一起,这两个步骤通常被称为亲核酰取代(看到帖子:亲核酰替换)
消除不发生除醛和酮因为离去基团必须极强的基地H(-)或R (-)。有利于酯也是合理的,因为离去基团RO(-)是相当的碱度的带负电荷的氧四面体中间。(注1]
- 但是等等!还有更多!步骤2后,我们有一个新的酮。我们以前见过,格氏反应迅速与酮在另一个加成反应(步骤3)。在这里,在步骤1中,我们形成碳碳和打破切断(π)。结果是一个三级醇盐(叔醇的共轭碱)。
(等等,你可能会问。如果我们只使用一个相当于格氏试剂,有可能使反应停留在酮阶段?简短的回答是“不”。(见注3长回答]]
- 最后,质子化作用这三级醇盐产量叔醇(步骤4)。
图形介绍:
6。摘要:格氏试剂的反应
它的关键的格氏试剂反应你会看到在大多数组织1和Org 2课程。
在下一篇文章我们将讨论另一种方法来搞砸了格氏试剂的形成,在这篇文章中,它涉及到反应。
在下一篇文章:保护组织格氏反应
笔记
注1:虽然醇盐(RO- - - - - -的共轭碱醇,pK一个16日至18日)不是任何人的名单上的离开团体,他们是一些25数量级比氢化物离开组(H -共轭碱的氢,pK一个40)和30多个数量级比烷基组(R -,烷烃的共轭碱,pK一个50)。因此,当醇盐的中间形成在步骤1中,没有任何深精力充沛的点球切断π键改革和RO -驱逐:毕竟,我们只是更换强碱(O)和一个类似的碱度。
注2。为什么酮比酯对格氏试剂反应吗?这就要求理解的现象π捐赠。氧上的孤对电子密度捐赠到羰基碳。这是值得一个单独的文章,但这是底线:
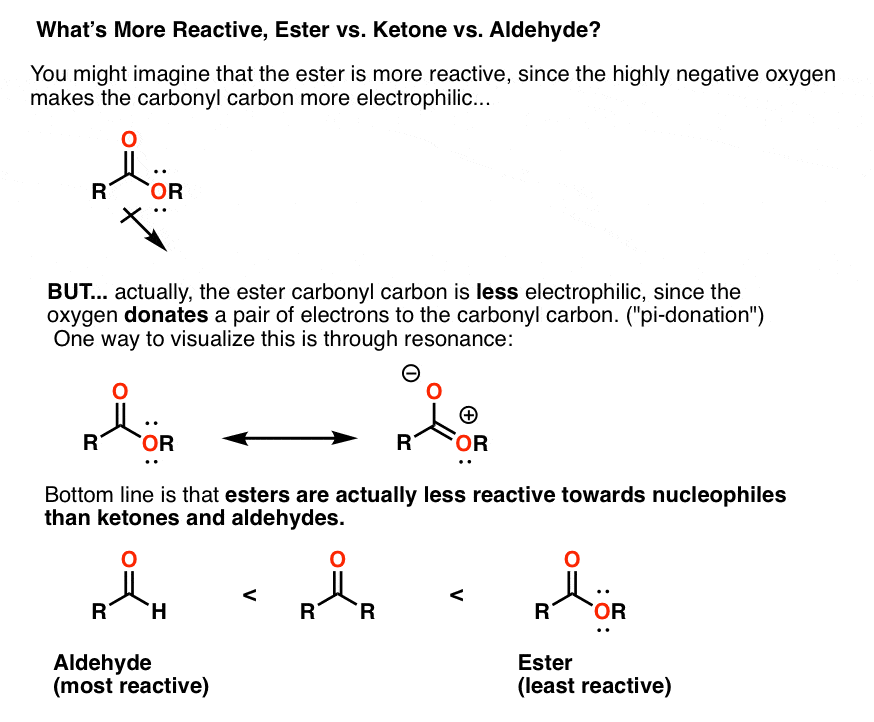
注3。唉,没有。使用1相当于格氏将导致0.5当量的叔醇酯开始和0.5的等价物。的原因是,步骤2[取消]非常快!
一旦消除发生时,我们会有酮酯的存在。有趣的原因(见注2)酮比酯对格氏试剂反应更大,这意味着他们将会更快地消耗。
(高级)引用和进一步阅读:
- 格利雅试剂
迪特马尔Seyferth
有机金属化合物2009年28(6),1598 - 1605
DOI:10.1021 / om900088z
历史概述在格氏试剂到教授迪特马尔Seyferth(麻省理工学院),《华尔街日报》的主编有机金属化合物。 - 二、三级Alkyllithium化合物和一些与他们互变现象的反应
亨利·吉尔曼,弗雷德·w·摩尔,奥格登Baine
美国化学学会杂志》上1941年,63年(9),2479 - 2482
DOI:1021 / ja01854a046
教授亨利·吉尔曼(爱荷华州)的先驱有机金属化学上半年的20个th世纪。在这篇文章中他描述的合成和反应各种alkyllithiums (n-butyllithium,年代-butyllithium isopropyllithium,t-butyllithium)。卤代烷的合成和锂金属,可以看到在实验部分。 - t-Butyllithium
保罗·d·巴特利特,c·加德纳情郎,罗伯特·b·伍德沃德
美国化学学会杂志》上1941年,63年(11),3229 - 3230
DOI:1021 / ja01856a501
这种沟通是一些传奇人物在有机化学和描述的制备t -butyllithium。 - 2-PHENYLPYRIDINE
c·w·埃文斯和c·f·h·艾伦
Org。Synth。1938年,18,70年
DOI:10.15227 / orgsyn.018.0070
这个过程的第一步是制备phenyllithium溴苯和金属锂。有机合成是一个有信誉的来源可再生的和独立测试有机合成过程。 - 锂-卤素交换反应的机制:一个文献之回顾
贝利,w·f·;帕特里夏·J·J。
Organomet。化学。1988年,352年(1 - 2),1-46
DOI:10.1016 / 0022 - 328 x (88) 83017 - 1
在现代有机化学,organolithium试剂很少准备从头开始(即使用李金属),由于唾手可得alkyllithium试剂从供应商(例如梅丽莎,BuLi试剂,PhLi,等等)。相反,这些试剂可以用于其他organolithium物种通过过程称为形式lithium-halogen交换。 - 这些锂试剂的什么?
汉斯·j·帝国
《有机化学》杂志上2012年,77年(13),5471 - 5491
DOI:1021 / jo3005155
汉斯教授帝国(美国威斯康星-麦迪逊)花了他的职业生涯研究organolithium物种的行为,这是一个他的研究和令人惊讶的发现他。这是经典物理有机化学。
你好,詹姆斯,
谢谢你创建这个网站。它帮助我很多,我不能感谢你才好。
我有一个疑问。请如果你能帮帮我。当你把一个位阻羰基或酸衍生衬底和同样位阻格氏试剂吗?机制的情况下我相信SN2和SN (addition-elimination)。反应继续进行吗?或不呢?或者如果真的那么如何?你有参考我可以通过吗?实际上早在大学,我的一个教授教我们关于笨重的广义相对论给予了氢化物,这攻击进一步位阻基质。但是因为,我不是在大学里现在,我不能问教授。 And I haven’t been able to find it in any standard textbooks. I checked clayden, klein and bruice.
你可能会有羰基的还原。我想看看3月的先进的有机化学。
如果不能用饱和氯化钠溶液在格氏反应的最后一步
格氏一般淬火与水溶液和有机溶剂提取。提取的目的是消除镁盐和其它极性的副产品。
生理盐水与CH3MgBr反应吗?
的问题之一氯化钠会溶解在适当的溶剂。但假设我们能够得到一些溶于醚,在CH3MgBr的存在。是唯一可能发生的一些格氏试剂的溴离子可能与一些Cl离子交换的氯化钠。这不会影响任何重要的格氏试剂的反应方式。我希望这回答了你的问题。
格氏试剂与烯烃反应吗?
谢谢你!
嗨,其他有机金属化合物的反应方式与格氏试剂相似吗?
不,不完全是,金属的选择可以在反应性有很大的影响。最相似的格氏试剂的试剂organolithium试剂,也将增加羰基(醛、酮、酯类)以及作为强大的基地。请注意,非常接近锂镁的电负性。一旦你向正确的一半的元素周期表(如铜或钯)反应变得截然不同;作为基本有机金属试剂不近,有更强的粘结烯烃等“软”的物种。
与酯反应,亲核试剂C2H5O——为什么不攻击在第二步中,只有格氏试剂攻击?
它可以攻击,但是攻击是可逆的。相比之下,与格氏反应是不可逆的自负碳离子是如此强大的基地和贫穷离开组织。
嗨
你能请告诉我格氏试剂反应1度,2和3度胺
没有反应,一般。
将th行动RMgCl /扶轮领导学院(RLi)对所有类型的酰胺?也回复多少摩尔的格氏试剂可以被一个摩尔的任何类型的酰胺(P / S / T)
我得到,但是我想要知道一个双键反应,如果在一个复合双键(2个碳)和醛,将格氏试剂与第一反应。
醛。格氏将增加共轭羰基(1、2)之前他们做加法(1、4)
更重要的是与格氏试剂反应,醛和酮和酯与烯烃吗
醛酮> >酯。
但这里有一个问题要问你。为什么?
Etheneone C2H2O。脂族结构与SP2杂交的碳氧双键。
啊,我从来没有听说过“乙烯酮”称为etheneone。还是应该加1,2。
乙烯酮与格氏试剂的反应是什么
我不确定你指的是乙烯酮,但如果你的意思是甲基乙烯基酮,答案是,格氏增加酮。
为什么需要垂直于羰基α碳酮的去质子化。
所以当碳氢键断裂,轨道(及其产生的孤对)与p轨道的C = O pi系统,允许共振稳定。
这样一个伟大的工作把所有这一切放在一起
谢谢菲利普,很高兴你觉得它有用。
这背后的原因是什么?
和醚做格氏反应吗?如果是的,那么如何?
不,他们不。这就是为什么醚是有用的保护组。
另一个问题!第一步是添加RMgX酮SN2吗?这是背后攻击吗?stereochem呢?
羰基的加成反应,它可能发生酮的脸。
你好!如何与格氏反应的二氧化碳形成carboxilic酸吗?任何职位呢?
什么是产品的立体化学反应中形成的醛或酮与格氏试剂吗
没有手性的来源,任何形式的另外一个手性中心将会导致立体异构体的混合物。
如何与腈、异腈格氏试剂的反应?
格氏试剂将酮,经过水解的中间亚胺。看到https://pubs.acs.org/doi/abs/10.1021/ja00796a022
相对反应活性顺序格氏试剂烷基
绝对的规模,它将与碱度关联很好,但这不是考虑空间因素。格氏通常不是他们的问题不是“足够反应”,但这衬底是位阻和副反应的结果。
当与格氏试剂反应2°酒精
这将是deprotonated环氧化物。酸碱反应,换句话说。就是这样。
好吧…我明白格氏试剂与所有上述化合物…但我困在这个问题…
乙酰溴化反应过度CH3MgI随后治疗的饱和溶液NH4Cl给? ? ?
你有一种酸氯化反应过度的格氏试剂。酸性氯化物与酯反应。格里纳补充道两次。
在许多情况下,检查一步说,“H +”。但是这可能会导致混乱,因为H +在叔醇的存在会导致形成碳正离子与醇(记住sn1)。所以H + NH4 + Cl -有时用于检查步骤。这是一个足够强大的酸(pKa 10)使质子化带负电荷的氧气,但不是一个足够强大的酸使质子化中性哦,因此SN1 / E1通路。
这有帮助吗?
嘿,什么是产品三个学位的醇反应“GR和H +的痕迹”。谢谢!
一个三级醇反应和H +“格氏试剂的痕迹”。这对我来说没有任何意义。格氏试剂H +所摧毁。
这是我的结论。这个问题在我的教科书。也许一个印刷错误。感谢所有的帮助:)
所有的氧气都含有化合物的反应活性顺序对格氏试剂(醛、酮、酯、酸氯、羧酸、酸酐、酸酰胺)?
酮醛和酸性氯化物> >酯> >酰胺。羧酸使质子化格氏他们不增加羧化物。
有可能形成一个与其他Gp2的格氏试剂金属?更大的Gp2金属离子不足够两极分化由于电荷密度较低?我只是跑考试复习课程,发现这个线程非常有用的一个不寻常的化学水平的话题。
你好,罗伯特。Organocalcium物种是已知的,但从我的理解往往给很多Wurtz-type耦合(就像organosodium试剂)。我还没挖到这个参考但介绍:j . Org。化学1993 58、1187。https://pubs.acs.org/doi/10.1021/jo00057a035
最终产品的立体化学是什么?
外消旋吗?
你好亚当,这将取决于几个因素。如果你谈论环氧开放,如果你从一个手性开始,non-racemic环氧,你最终会得到一个手性,非外消旋产品由于异构中心未受影响。如果你在谈论一个非手性的醛或酮,然后没有任何影响手性你会获得一个外消旋的产品。如果你在谈论一个手性之外,但外消旋的醛或酮无手性影响你最终会得到一个非对映体的混合物。这是一个很好的问题,值得自己的博客。
你怎么知道何时使用H3O + v . NH4Cl ?
这是个很好的问题。因为你可能会看到我把。很依赖老师。
介绍有机的目的,然而,你可以认为他们是相同的。
在实践中NH4Cl有点温和,尤其是acid-sensitive可能与H3O +反应的分子。NH4Cl是一个“安全”的选择,当你需要一个温和的酸检查。
反应性的顺序正确吗?格氏反应顺序:氯>酐>醛>酮>酯>酰胺?
嘛。我不想尝试运行之间的竞争实验酸氯醛,但醛反应肯定比酮。
能与格氏试剂反应没有官能团的分子,如methylcyclopropane ?
Methylcyclopropane,不,因为没有电子的负碳离子会下沉。他们可以与酸性末端炔烃和环戊二烯碳氢化合物反应。
你好,为什么不能格氏反应的α碳酮和醛的pKaα碳约20 ~虽然烷烃的pKa氢~ 50 ?
很好的问题乔恩。之一,我们不要提太多棘手的部分是为了使碳氢键是酸性的,它必须是一致的羰基在90度。所以当格林尼亚附近的羰基,两件事可能发生;它可以添加,也可以删除碳氢键。但是就像我说的碳氢键必须完全一致,而羰基是没有这样的限制。
其结果是,而且往往是比去质子化在这种情况下,只要羰基不太位阻。
原谅我如果我错了,但是出现的插图略高于2头”。与醛和酮格氏反应,”做了乙基(飞机的)环庚烷的甲基环改变错误?
是的,你是正确的。谢谢你的位置。固定!
所以我有一个问题,为什么试剂不会在酮而不是进攻羰基质子化了的吗?
试剂的情况下能使质子化格氏或酮位阻。并发症之一的酸性α位置醛或酮的α碳氢键必须与C = O债券为了它是酸性的;否则由此产生的负碳离子共振稳定。没有这样的障碍除了羰基。
格氏酰胺?
当然不是为主要或次要酰胺。介绍有机化学puposes,通常酰胺认为是惰性…。但谷歌“发现酰胺”更先进的治疗。
我想知道关于不同格氏试剂的反应,用同样的R组。意思我,Cl, Br。如果我有两个甲基卤素的解决方案,哪一个会更容易形成了格里纳?
卤的顺序活动是我> Br > Cl。看到3月先进的有机化学第五版805页。在“Metallo-de-halogenation”。
NH4Cl或H3O +的影响与GR环氧的sn2机制?
NH4Cl或H3O +将摧毁格氏试剂。一些路易斯酸(例如CeCl3)与格氏然而兼容。
比酮酯活性较低,碳酰卤化物呢?
酯肯定是不如酮反应。我不知道一个好的竞争实验碳酰卤和酮或醛。我想说的是,在这三个案例第一而且非常快。
一个问题:如果一个等效的格氏试剂反应的化合物,其中包含一个双键和羰基(C = O),格里纳就会攻击哪一个?
羰基。除了双键可以影响如果你添加一个催化量的铜(I)盐CuBr等。
格氏试剂的反应会受到影响的变化从质子,质子溶剂。
格氏试剂将被摧毁,如果添加到质子溶剂如酒精或水。
嘿如果1,3 dibromopropane与格氏反应吗?
你会得到一个最有趣的产品C3H6化学公式。你能算出它是什么吗?
格氏试剂和氨之间的反应是什么?
我从来没有见过在氨格氏试剂的使用,尽管有时叔胺被添加到格氏帮助他们溶于苯或甲苯。