芳香分子的反应
分子内的傅克反应
最后更新:2022年10月7日|
分子内的傅克反应
在这篇文章中我们探索分子内的傅克反应。但首先,快速复习在分子内反应。
表的内容
1。快速回顾:分子内反应可能会非常棘手
这是一个即时的公式一个有机化学考试的问题。
先从一个简单的反应,学生理解相当好,像威廉森醚合成…。
(奇怪,一个好的目的)
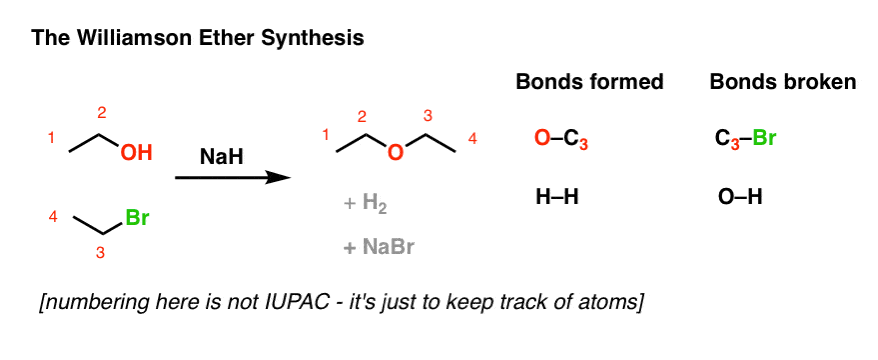
…现在,做一个简单的修改通过添加一个键。瞧。即时难题!
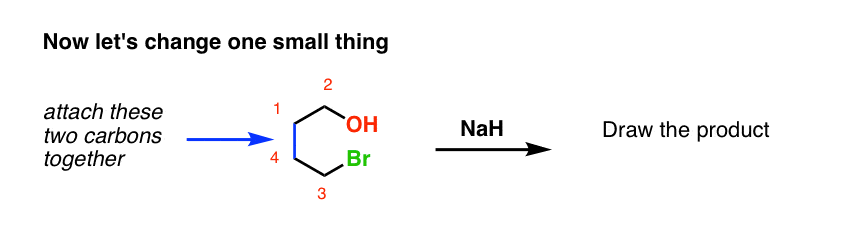
上面的第二个问题是一个一个的例子分子内的反应,亲核试剂和亲电试剂在同一个分子,他们的反应的结果是形成一个环。在过去的帖子在这个问题上,我使用了类比的皮带bdapp平台。它仍然有效。
{kind=link}
为什么是分子内反应好考试问题吗?因为他们解决学生学习记忆的反应一个简单的例子,表和那些知道(最重要的是,可以申请!)债券和债券形成的模式打破了。此外,它不涉及新概念,这使得它完全公平的游戏。
分子内的变异存在很多不同的反应,出现如此频繁,值得一提的是分开。的傅克烷基化和傅克酰化反应也不例外。
2。分子内的傅克烷基化
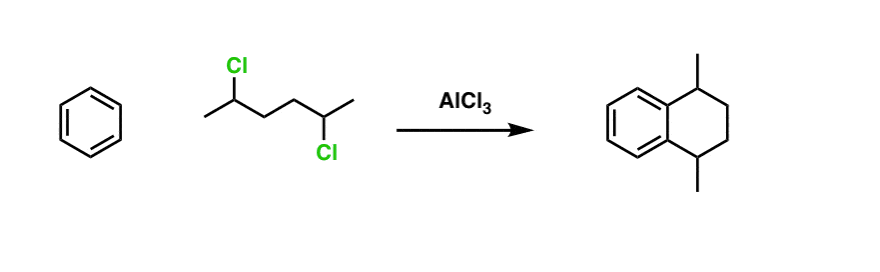
这是分子间傅克烷基化的一个例子。的亲核试剂芳香环,亲电试剂是烷基氯。添加一个小(AlCl催化剂3)和繁荣!亲电芳香取代反应。
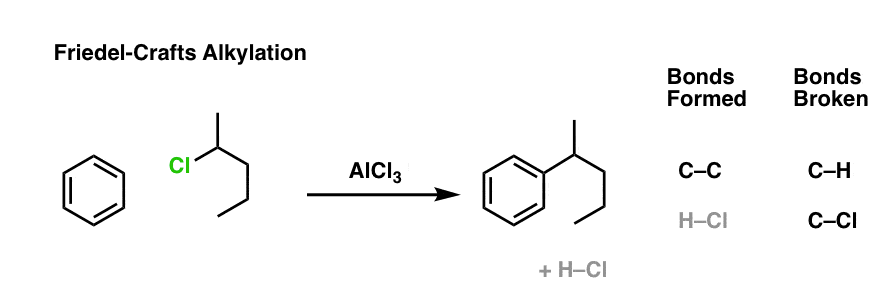
现在让我们改变一点点。我们会把烷基卤的环通过一个新的碳碳键,然后添加催化剂。是什么产品?
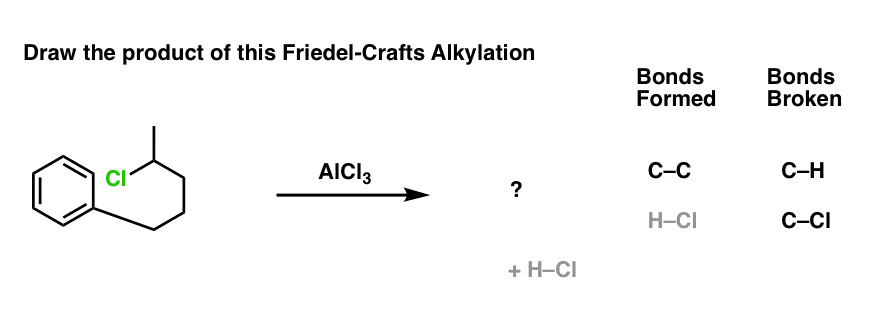
没有新概念!相同的债券形式和破坏模式。但如果你还没有见过这样的一个例子,它可能把你完全记住笔记。这是计划!(“邪恶笑”的提示记录)。
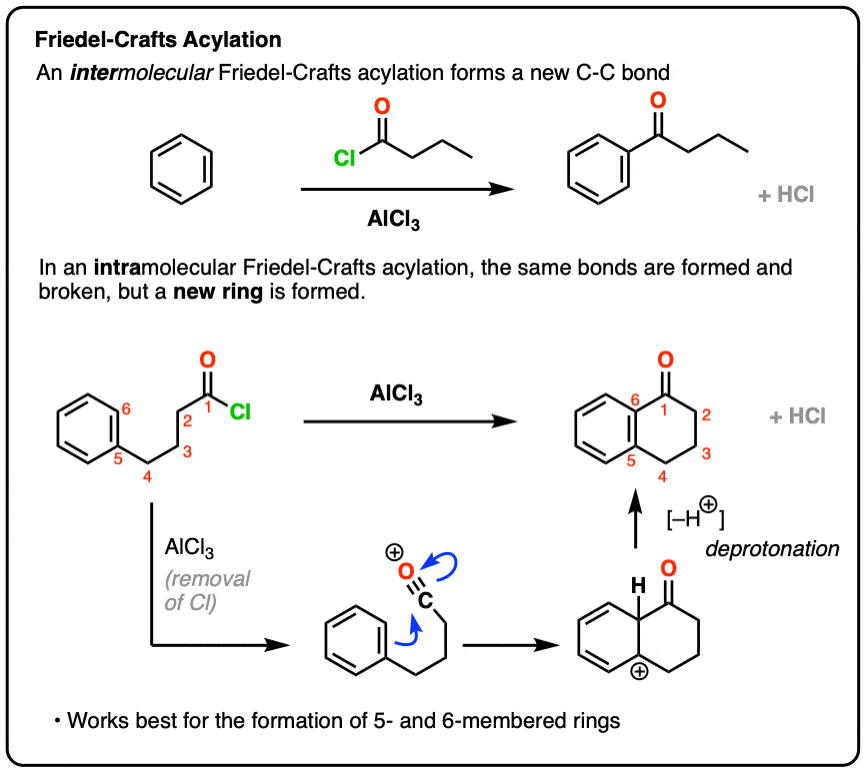
好的。这是机制,下面画出最终产品。
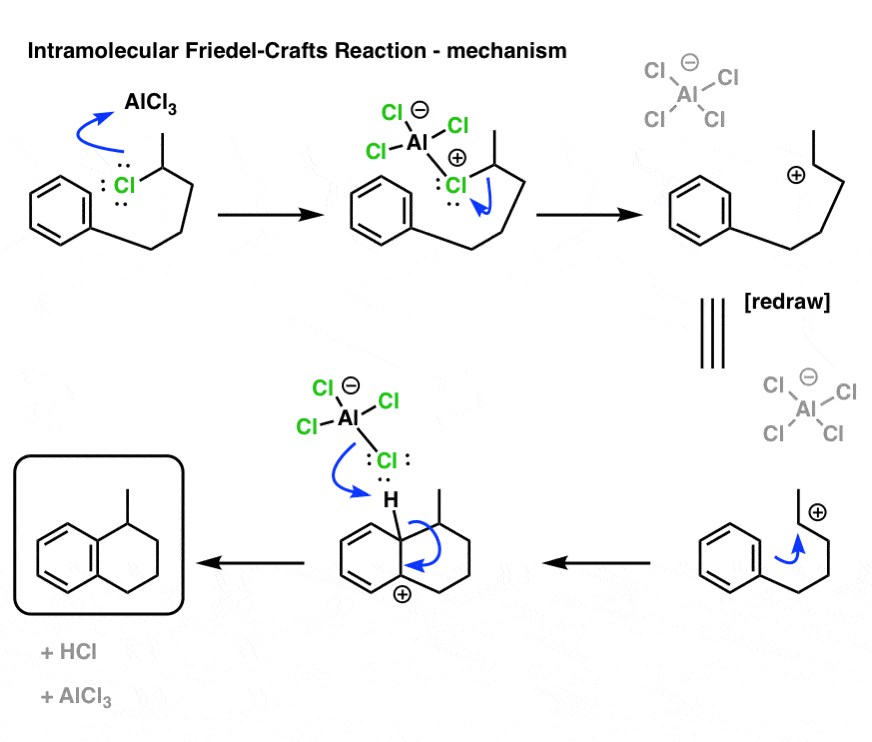
我想让你们注意,整个模式的债券和债券形式,打破完全相同的在分子内的情况下分子间的情况,即:形成碳碳和氯,碳氢键和C-Cl划清界限。
永恒的建议画出这样的反应机制:
- 碳原子数!很简单“下降”碳在你的图纸,这将失去你点。
- 首先画出“丑陋的版本”,然后先让它看起来不错。不要担心使它看起来更漂亮,直到你有了债券形式和休息。
的傅克反应,分子内的版本最适合做六元环,但也可以使用傅克反应使5 -和7 -元环(没有显示)。
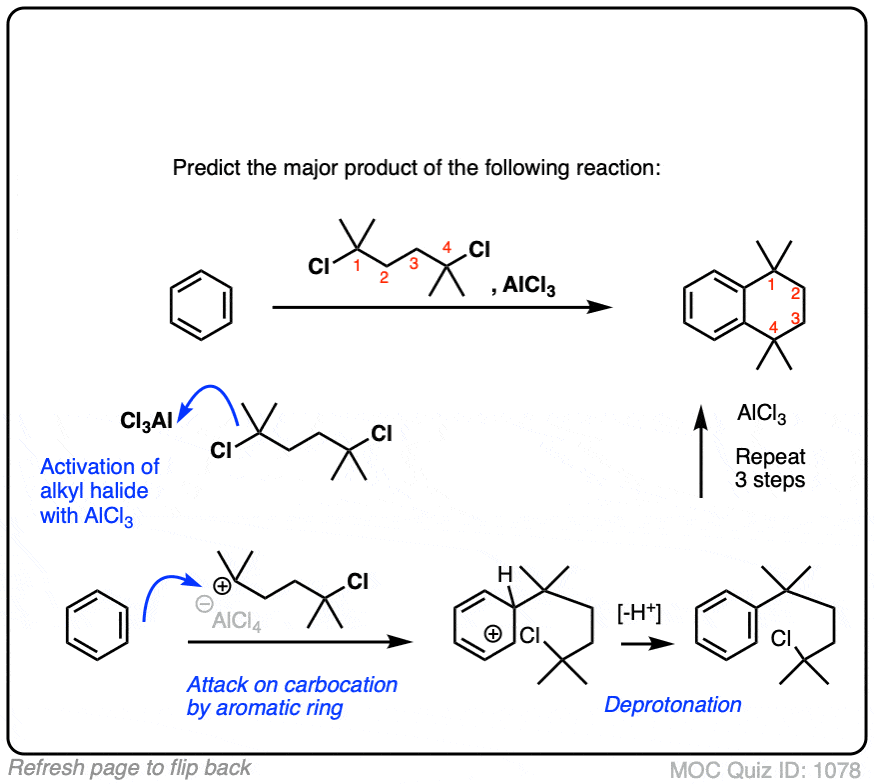
这是一个更先进的实践问题,始于苯。你能画出最终产品吗?(下面的回答)
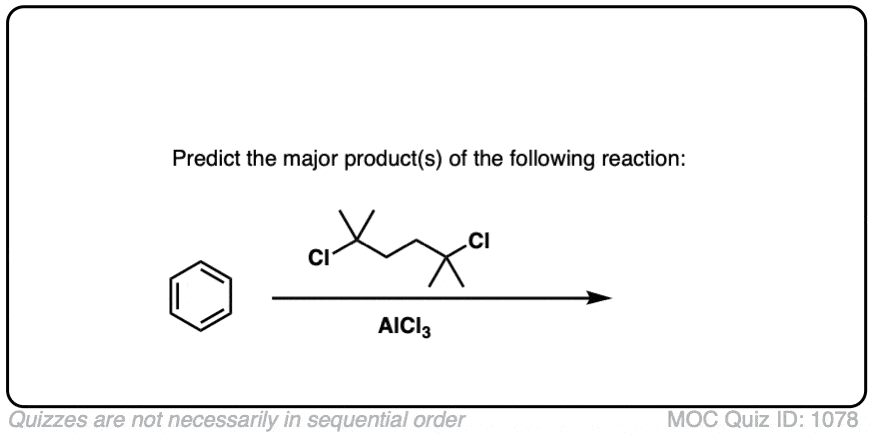 点击翻转
点击翻转
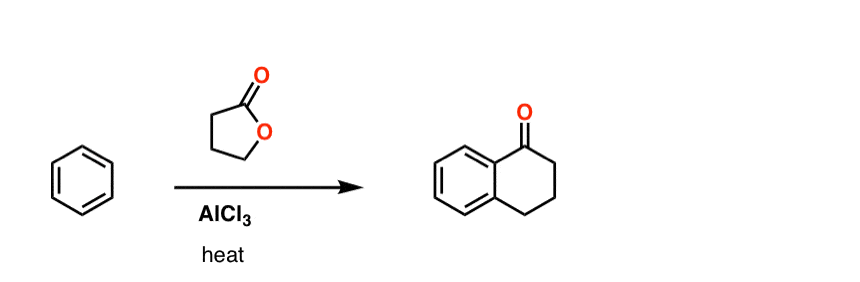
3所示。分子内的傅克酰化
一旦你看到了分子内的傅克烷基化,分子内傅克酰化并不会感到惊讶。
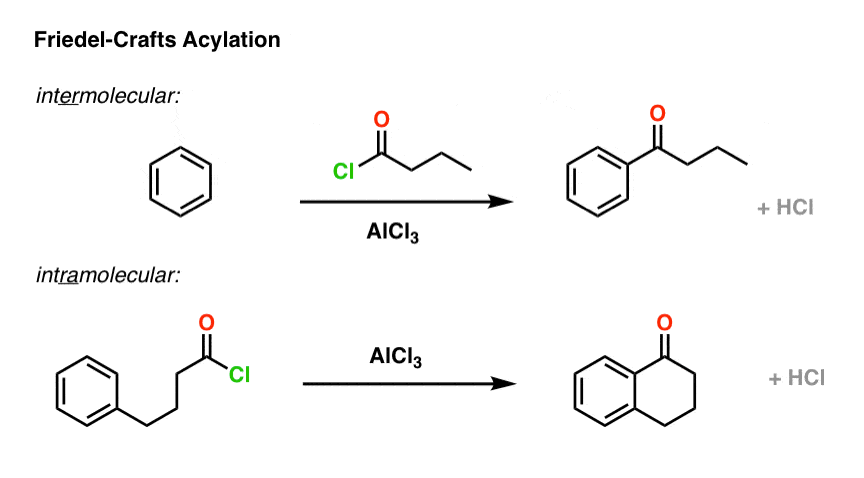
再一次,我想让你确认债券形成和破碎的每种情况是完全相同的。唯一的区别在于,在第二种情况下,亲核试剂和亲电试剂通过系绳连接到对方。
你可能不会看到一个小皱纹,但到底。我们曾经见到过的酰卤化物(酐)的傅克反应,但对分子内傅克酰化一件有趣的事情是,羧酸也可以参与。例如,治疗羧酸与强酸如H2所以4结果在一个分子内傅克酰化:
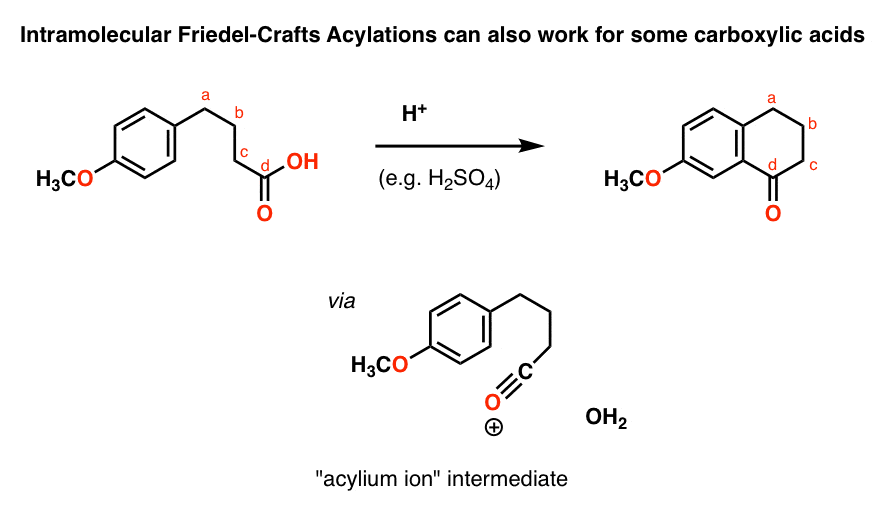
原因之一这适用于内部和分子间的亲核试剂是那么紧密亲电试剂。这同样的效果的浓度亲电试剂是急剧增加。你可能会看到其他的例子,你可以使用一个贫穷的“侥幸”亲核试剂(或亲电试剂)如果是在一个给定的反应在一种分子内的方式完成的。
这是一个挑战的问题。你能画出这个序列的傅克反应的产物吗?
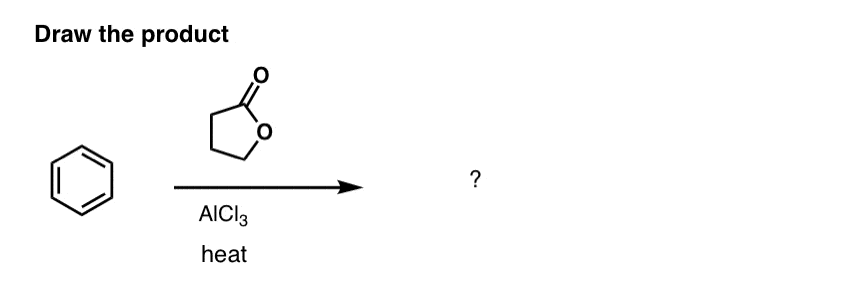
(下面的回答)
4所示。摘要:分子内的傅克反应
没有真正开创性的新概念,但总是有助于保持警惕分子内的反应的例子。这个概念永远不会消失。你会看到一遍!
在本系列我们在下一篇文章中我们将讨论一个完全不同的类置换反应的芳香族化合物,适用与electron-poor芳香组。它被称为亲核芳香取代反应。
笔记
注1。重排和环关闭。我们已经见过主烷基通过氢化物(和卤化物可以重新排列烷基)转变为更稳定的碳正离子中间体。所以当一个主烷基卤化物参与分子内的傅克烷基化反应吗?重排首先发生,或环闭合速度?
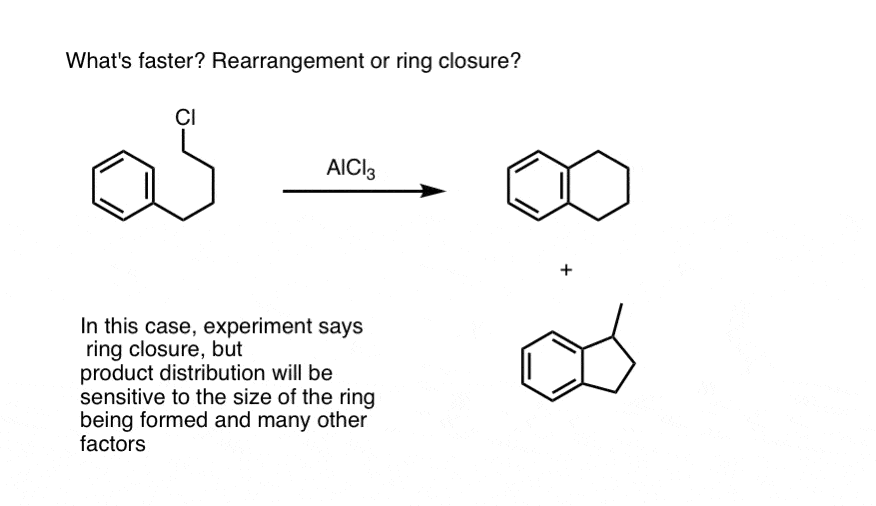
这不是你能回答的问题简单地思考这个问题。当有竞争反应速率,确定的唯一方法通过实验。
这是研究,论文全文的重排和环闭合率的研究在这里。[J。Org。1966年化学,31日,89]。表明关闭六元环比关闭快到5元环。
{kind=link}
(高级)引用和进一步阅读
傅克酰化和烷基化分子内,这是有用的双环或多环化合物的合成。
分子内的烷基化:
制备的分子内fc烷基化是最成功的六元环,虽然5 -和7-membered戒指也以这种方式被关闭。它比5人比较容易形成六元环的反应。4 -苯基1-butanol给出了环化萘满在磷酸50%的收益率,而3 -苯基1-butanol主要是脱水烯烃。
如果一个潜在的碳正离子中间体可以进行氢化或烷基转变(或Wagner-Meerwein重排),这发生在偏好环关闭5环。这反映了一个普遍的趋势6 > 5,7在环分子内关闭的傅克反应。
- 新傅克反应化学。XVI.1复议Cyclialkylation和竞争反应的某些Phenylalkyl Benzoylalkyl, Acetylphenylalkyl氯化物
阿里阿里夫和罗伊斯顿·m·罗伯茨
《有机化学》杂志上1966年,31日(1),89 - 95
DOI:1021 / jo01339a018 - 新傅克反应化学。第十九。一些phenylalkanols Cyclialkylations
阿里夫和罗伊斯顿·m·罗伯茨
《有机化学》杂志上1969年,34(11),3571 - 3574
DOI:1021 / jo01263a075 - 傅克反应cyclialkylations某些mono -和diphenyl-substituted醇烷基氯化物
阿里阿里夫和罗伊斯顿·m·罗伯茨
《有机化学》杂志上1972年,37(26),4227 - 4235
DOI:1021 / jo00799a001 - 环化2 - [N - (methylsulfonyl)苯胺基]乙醛二乙酯缩醛树脂吲哚。立体效果的证据在分子内的亲电芳香取代反应
理查德•j•桑德博格和约瑟夫·看
《有机化学》杂志上1984年,49(2),249 - 254
DOI:10.1021 / jo00176a007
本文有一节讨论不同的过渡态的几何图形为5 - 6元分子内圈关闭。5人过渡状态更紧张,尤其是如果它是假定碳亲电攻击一个方向垂直于这个平面的戒指。进一步的例子分子内的傅克烷基化: - 实验针对总萜烯的合成。第十七章。五环三萜烯的合成方法的发展基于机械的傅克反应的立体化学的结果的解释cyclialkylation反应
罗伯特·e·爱尔兰,史蒂文·w·鲍德温和史蒂文·c·韦尔奇
美国化学学会杂志》上1972年,94年(6),2056 - 2066
DOI:10.1021 / ja00761a044
这的一个例子是一个环多环系统,b的产品是一个3:1混合的地方:一个甲基异构体在新环结,反映出偏爱在过渡态组织的方向。 - 系统的研究苄阳离子进行环化反应
Steven r角和迈克尔·s·路易
《有机化学》杂志上1991年,56(8),2853 - 2866
DOI:10.1021 / jo00008a049
本文讨论了分子内fc烷基化到苄基的位置。六元环形成更有效地比5 -或7-membered戒指。 - Enantiospecific合成(+)- 1 - (R)苯基3-methyl-1, 2, 4, 5-tetrahydrobenz [d] azepine (+) - (S) -N-methyl-1 -苯基乙醇胺(halostachine)通过芳烃铬tricarbonyl方法
史蒂文·j·库特斯蒂芬·戴维斯,大卫Middlemiss,艾伦·内勒
四面体。1989年,30.(27),3581 - 3588
DOI:10.1016 / s0040 - 4039 (00) 99447 - 4
分子内的fc烷基化完成标准条件(如H2所以4/组织或住宅4渗出性中耳炎?2)导致外消旋的产品,如预期。手性可以通过一个不寻常的(但先进)诱导方法包括有机金属化合物,可逆地形成一个arene-Cr(有限公司)3复杂。分子内酰化: - 2-Hydroxy-17-equilenone的合成
巴赫曼和w·j·霍顿
美国化学学会杂志》上1947年,69年(1),58 - 61
DOI:10.1021 / ja01193a014
一个经典的分子内环合试剂phenalkyl羧酸是PPA(多磷酸),这可以通过添加P2O5和磷酸。这是不习惯这一切了,因为这是一个疼痛的处理——非常腐蚀性和粘性。 - 甲磺酸。一个有用的使环化酸性试剂
阿尔贝托·a·莱昂,圭多涂抹,即罗伯特·西尔弗曼
《有机化学》杂志上1984年,49(23),4544 - 4545
DOI:10.1021 / jo00197a047
MSA(甲基磺酸,CH3所以3H)是一种另类的PPA fc环合反应。它是便宜的,现成的,是一个容易处理的液体,可比PPA的酸度。 - 光谱和其他属性的大型环Mono -二聚的Benzocyclanones由高度稀释傅克反应
m·舒伯特w·a·斯威尼,h . k . Latourette
美国化学学会杂志》上1954年,76年(21),5462 - 5466
DOI:10.1021 / ja01650a060
而分子内fc酰化主要用于关闭5 - 6 -,和7-membered环,更大的戒指可以封闭高度稀释技术。 - 有效的选择indenones合成
发达的肌肉。弗洛伊德和乔治·罗杰。艾伦
《有机化学》杂志上1970年,35(8),2647 - 2653
DOI:10.1021 / jo00833a036
分子内fc酰化与酰氯/ AlCl也可以做3.Tandem酰化和烷基化反应也可能;这些都是称为cycli-acyalkylations。不饱和酸内酯也可以被使用。 - α-TETRALONE
塞西尔·e·奥尔森,阿尔弗雷德·r·巴德·g·达纳·约翰逊
Org。Synth。1955年,35,95年
DOI:10.15227 / orgsyn.035.0095
第一个程序,提交的a·r·巴德使用内酯有效地做一个串联fc cycli-acylalkylation。a·r·贝德发现了奥尔德里奇的化学公司,这使他非常富有,永远改变了化学研究的化学家现在不需要花费他们的努力重新合成了常见的起始原料,随着这些现成的可用在高纯度。 - 霍沃思合成的范围
以色列Agranat Yu-Shan施
《化学教育1976年,53(8),488年
DOI:10.1021 / ed053p488
霍沃思合成的第一部分涉及fc酸酐酰化,其次是分子内fc烷基化后减少酮。
无水三氯化铝在回答我想知道激活内酯,# 2是哪一部分的内酯和三氯化铝反应吗?内酯环的氧或氧羰基吗?我该如何选择其中两个?
就像酰氯氯与三氯化铝反应
好问题。三氯化铝的羰基氧被激活。这个设置时作为离去基团的羧酸盐在sp3杂化碳环的攻击。这是第一个傅克反应。第二个弗里德工艺是一种酰化反应,O-AlCl3离去基团(-)。
我最通过四氢萘酮机制的方式,但我不知道最后的分离铝复杂。我的桥氧酯复杂最初三氯化铝第一,破环和acylium形成。fc酰化和rearomatization正常,但这里是我困惑的地方。中心带有一个负电荷,铝和氧气不再是带正电荷,所以如果切断键断裂烷基化物,我有一个双重负面铝物种。我到底哪里错了?
谢谢!
正确的。首先尝试烷基化将RCO2Alcl3——你的离去基团,这对FC酰化将吃得饱饱的
先生请告诉机制当酸酐与2情商的三氯化铝与苯反应
答案# 2 ? ? ?我需要一个解释! ! !
这是一个序列的傅克酰化然后傅克烷基化。第一步:内酯的三氯化铝与氧气反应,导致形成acylium离子的芳环的攻击。现在你有一个氧气协调三氯化铝(一个好的路易斯酸)。三氯化铝把氧气转换成一个好的离去基团。所以接下来会发生什么是攻击的芳环碳轴承氧气,导致一个新的六元环的形成。
在示例中给出内部摩尔佩珀freidal工艺反应,acylium
阳离子攻击元邻对位甲基分组指导,主要产品。
你提议,七元环会形成包含反式双键。这不是一个稳定的分子。
这篇文章很好。我想知道您具体对源或参考例子分子内的羧酸酰化我有一些困惑为什么没有水解醚。
底部的参考链接,但这是有机合成,科尔。4卷,p。898 (1963);35卷,p。95 (1955)。
酒精会发生什么是最终坐标向攻击三氯化铝激活它的芳环,它作为离去基团离去。
但R组是一个邻对位董事和主要产品是帕拉,为什么不是它关闭,连接的对位芳环吗?
答案是,帕拉产品会非常紧张。羰基不能达到对位的方式导致non-strained键长和键角。如果你不确定关于这个我邀请你,试图建立一个模型!
在实践中问题# 1看来你改变反应物从七个碳链的实践问题六碳链关键的答案。
开枪。谢谢你柯克,将修复。
固定的,最后。
我怀疑会有任何接受者提出四氢萘酮形成机制通过上述傅克反应:)我曾经把一个在考试中像3或4年了。学生没有欣赏的挑战…
是的,那绝对不会赢得任何球迷。