芳香分子的反应
更多的反应芳香Sidechain:减少硝基和拜耳威利格
最后更新:2022年10月15日|
在这里,我们涵盖三个新反应的芳香族取代基:
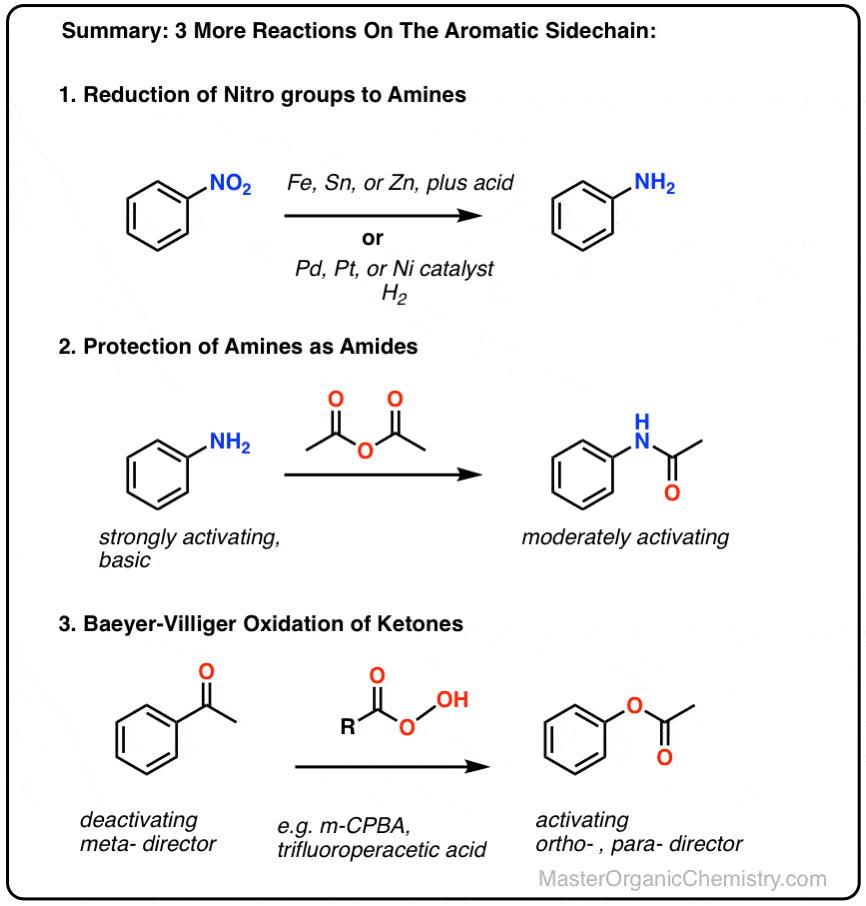
表的内容
- 三个反应的芳香族取代基
- 减少硝基
- 氨基的保护
- Baeyer-Villiger反应
- Baeyer-Villiger反应机制
- Baeyer-Villiger迁徙的能力
- 提醒关于混合的“酰胺”和“酯”
- 摘要:还原硝基和Baeyer-Villiger反应
- 笔记
- 测试你自己!
- (高级)引用和进一步阅读
1。三个反应的芳香族取代基
芳香族化合物的反应在本节中,我们看到两个重要类的反应:
- 首先,有芳香取代反应,债券的形式,直接在芳环上,导致新的取代基。我们看到在亲电芳香取代,例如[看到帖子:介绍亲电芳香取代反应]
- 第二,所有的取代基的反应芳香环。我们覆盖的减少金属羰基合物亚甲基(CH2在以前的文章)碳(链接),以及氧化苄基的碳氢键C-Br和切断债券(链接]。
取代基的反应是一个重要的部分,因为修改了芳环取代基可以显著改变发生亲电芳香取代反应。看到的:理解邻、对位、和元董事]
例如,我们看到,减少C = O CH2有巨大影响的反应环,因为它将一个安静下来吗元——导演激活昊图公司- - - - - -,帕拉——导演。
在这篇文章中,我们将结束这个话题有三个反应的芳香族取代基,包括:
为什么这些反应重要?
学习的最终目的反应是能够应用它们合成分子的我们自己的设计更简单,更便宜的组件。每个反应是一个“工具”,是一个特定的目的允许某些特征的形成和断裂的债券。工具越多你的“商店”,权力越大设计合成的有机化合物。
2。减少硝基
其中一个最重要的芳香族取代基的反应减少硝基团体胺。这可以通过使用两个通用方法:
- 添加一个容易氧化金属如铁(Fe),锡(Sn)或锌(锌)的酸,如盐酸(但往往只是写,“H +”)不会转换2对NH2。
- 加氢在钯、铂、镍催化剂也没有进行转换2对NH2。
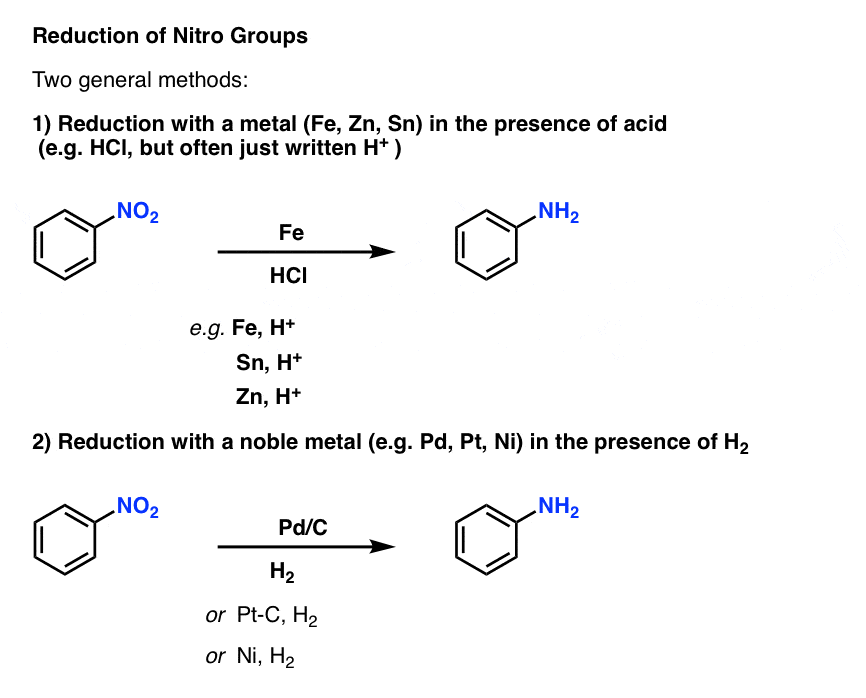
方法是有效的,可以被认为是本质上是相同的对于我们的目的。[机制呢?(注1]
用催化氢化作用轻微的优势之一是,它可以在中性pH值,因此不会影响acid-sensitive官能团。
3所示。氨基的保护
最重要的一个硝基的还原特性胺是它将强,才会安静下来元导演取代基强烈激活,昊图公司- - - - - -,帕拉- - - - - -指挥取代基。
事实证明,然而,这实际上可以介绍一些新的问题!
- 首先,氨基激活,亲电芳香取代反应可以发生不止一次,但是多个次,导致不受欢迎的产品。
- 其次,上的孤对胺是基本。反应需要Brønsted或路易斯酸催化剂(如傅克反应、sulfonylation或硝化)不加速的反应亲电试剂,而是导致协调的酸胺孤对!
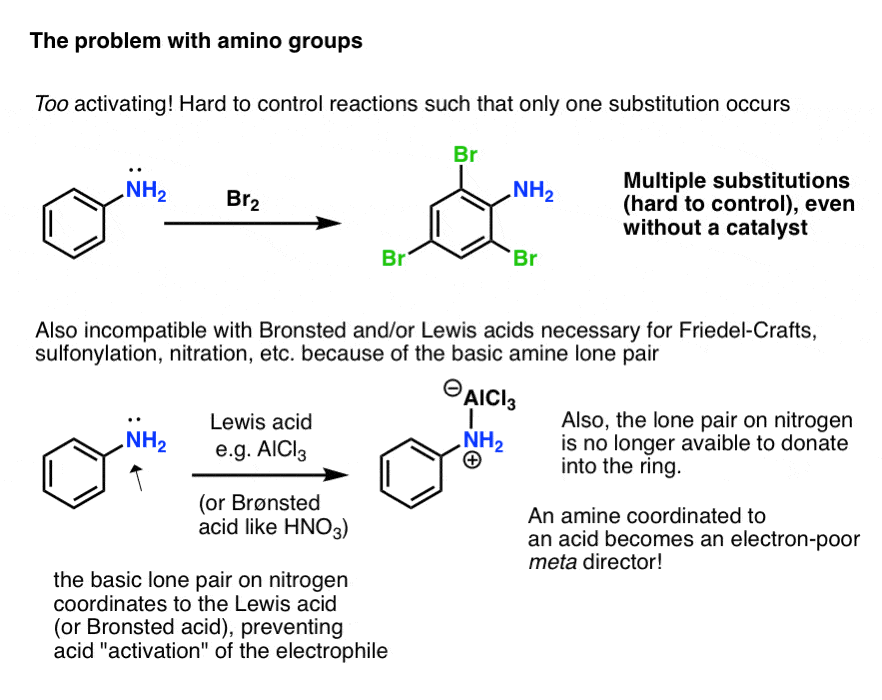
此外,这意味着氮上的孤对不再能够捐到芳环(通过“π捐赠”)。因为氮的电负性大于碳、氮酸协调是吸电子和实际行为元——导演!
那么我们如何驯服的“野马”是一个免费的氨基取代基?
幸运的是,它是相当容易的。一个常见的方法是将自由胺成一个酰胺试剂如乙酸酐(Ac2O)。
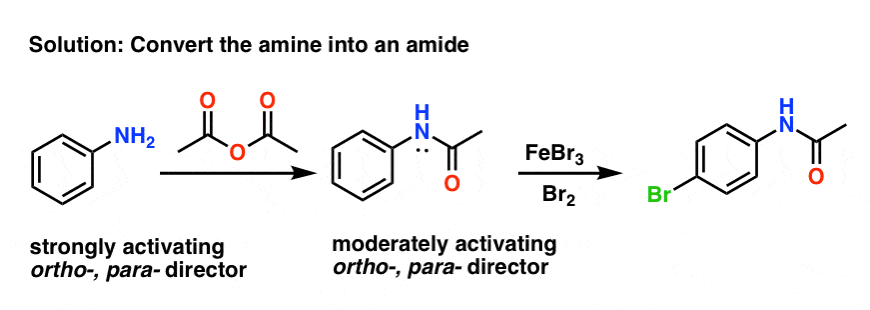
结果仍然酰胺昊图公司- - - - - -,帕拉董事(注意,孤对氮!)但不是那么激活免费胺。
此外,酰胺更兼容布仑斯惕和路易斯酸比免费胺。
如果空闲胺需要之后,它可以通过对酰胺酸性水解(水,Hbdapp平台2所以4、热)。
4所示。Baeyer-Villiger反应
另一个有用的反应对于我们的目的,不限于芳香酮是Baeyer-Villiger反应。治疗的酮与peroxyacid像米-chloroperoxybenzoic酸(m -CPBA)或trifluoroperoxyacetic酸的结果在一个氧原子的重排反应之间插入了芳环和碳羰基碳,将酮成一个酯:
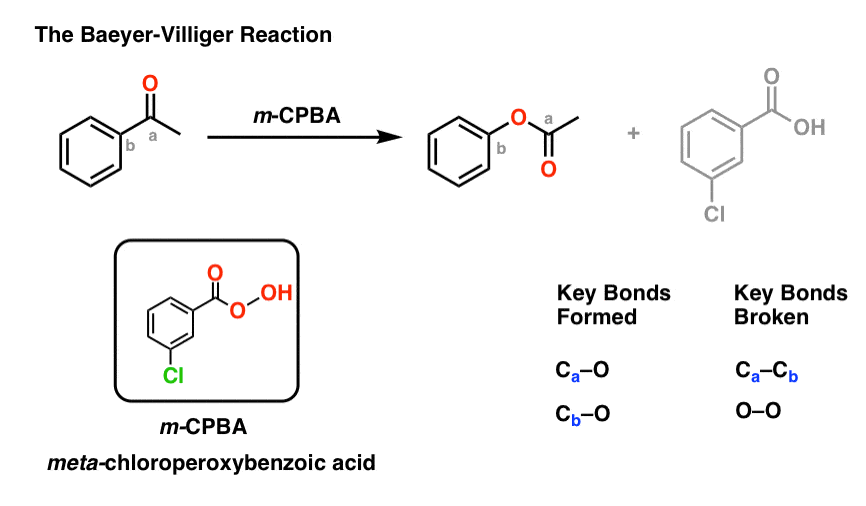
(不,“典型的”像KMnO氧化剂4铬酸或PCC将不这样做。有具体原因peroxyacids工作!)
Baeyer-Villiger反应填充一个重要的差距在我们的合成方法的“工具箱”。
到目前为止,我们已经学会了如何形成碳碳,氮,C,和C -(卤素)债券芳环,但不切断。
也注意Baeyer-Villiger,如硝基的还原胺处于待发状态转换,元导演羰基组一个激活,昊图公司- - - - - -,帕拉- - - - - -导演氧(氧上的孤能够pi-donation)。
现在有趣的部分。它是如何工作的呢?
5。机制Baeyer-Villiger
在Baeyer-Villiger的第一步,一个氧气从peroxyacid攻击酮,导致一个四面体中间。质子转移然后导致关键中间体:
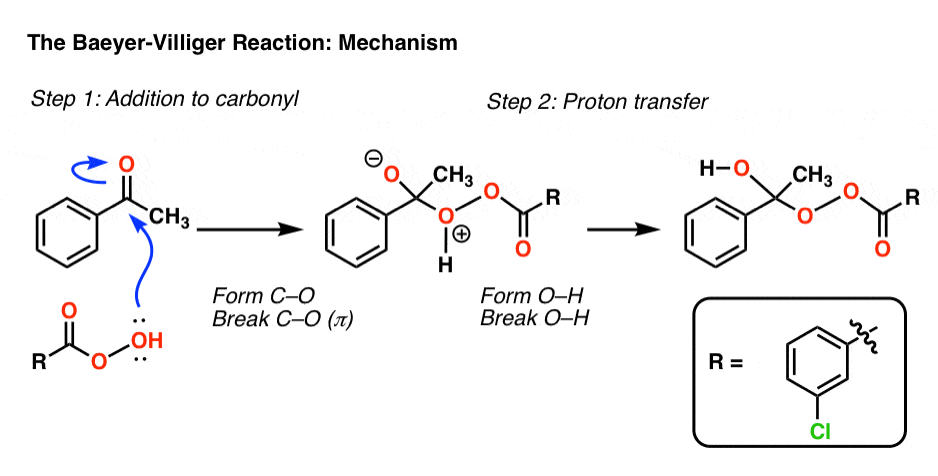
现在有重组的关键一步。在这个步骤中,一个σ键作为亲核试剂,攻击氧气和打破了o - o键弱(约35千卡每摩尔)。
看看你是否能按照箭头在这一步:
- 在第一弯箭头,碳碳优惠和切断形式
- 在第二曲线箭头,oo的断裂
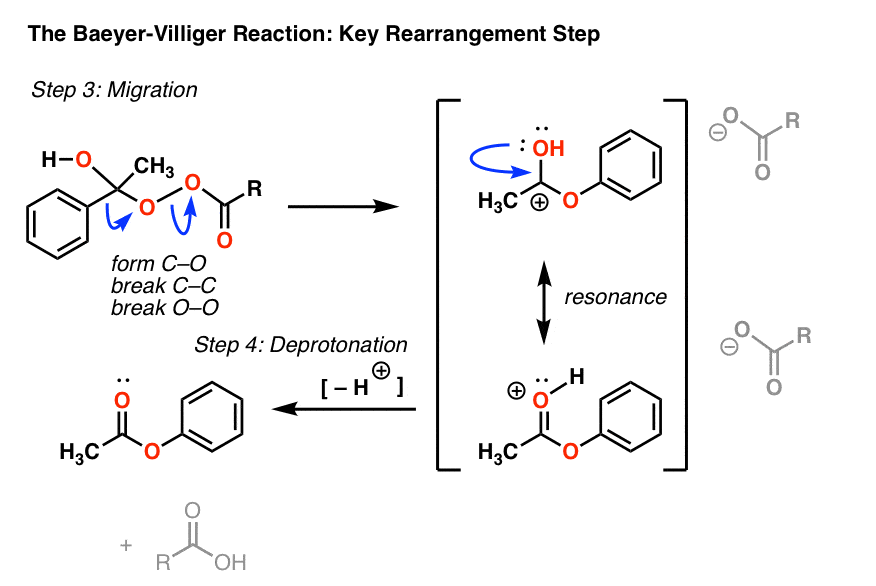
打破了碳碳键导致碳正离子。我们可以画一个共振形式的孤对氧气使一个新的碳给一个质子化了的π键酯(第三个弯曲的箭头)。(注:也可以画出这个同时发生重排,见注2为例]
这是然后deprotonated(由共轭碱酸)给最后一个酯产品和m -氯苯甲酸。(不再过氧化苯甲酸,o - o键!因为它失去了)
在这种重排反应,我发现它有用的画“丑陋”版本的产品,正确的连接,但不佳。徘徊在这里“丑陋的版本”的形象(链接到图片)
{kind=link}
6。迁徙Baeyer-Villiger氧化的能力
好问题!这是一个很棒的问题,回答会有点超出了标准的课程材料,所以我会解决这个问题注3]
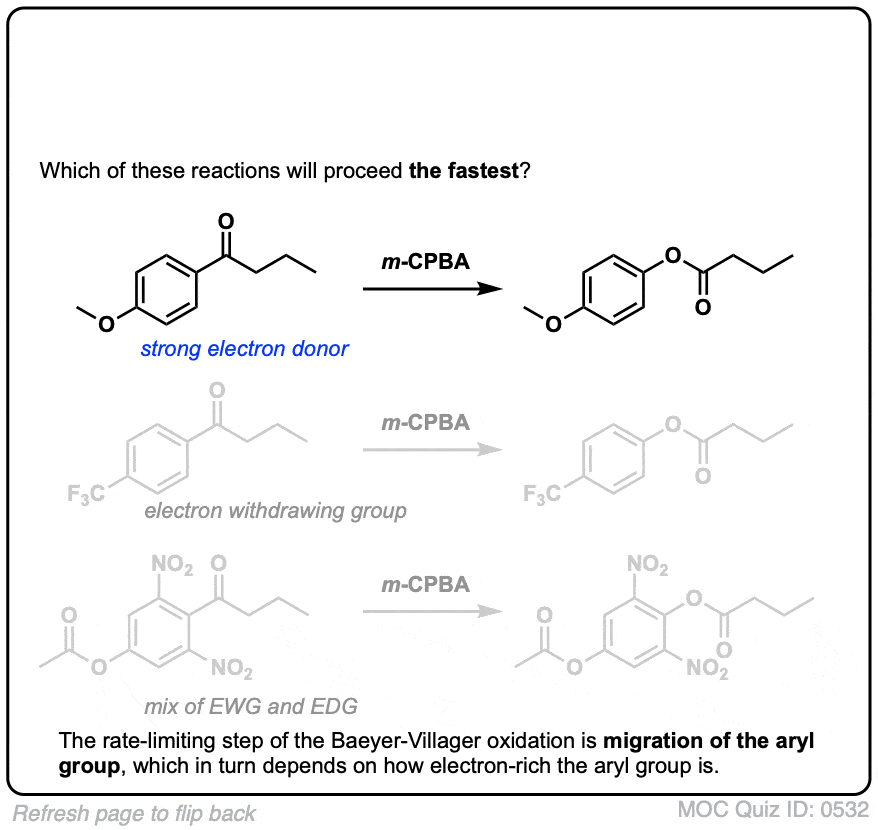
在简短的解释,有一个普遍趋势迁徙能力和碳正离子的稳定性(之间最大的例外苯基)。
迁移能力的一个大致顺序是:叔碳(最快)>仲碳,芳基(例如C6H5)>主要碳>甲基(慢)。
(后来在(胺),你可能会遇到贝克曼,霍夫曼,库尔修斯,和施密特重组发生通过类似的迁移步骤)。
倒叙:Hydroboration-Oxidation,σ键作为亲核试剂
在我们以前见过这样的重组步骤吗?
我们遇到的三个类亲核试剂在有机化学看到的:的三个类亲核试剂(孤、π键和σ键)σ键的亲核试剂是迄今为止最棘手的。之前,我们已经看到σ键等亲核试剂的情况下:
- 1,2-hydride转变(碳正离子重排)
- 烷基转移(碳正离子重排)
- 的氧化步骤在硼氢化反应
这最后一个例子是最相关的。
您可能还记得,在氧化硼氢化反应,过氧化氢离子攻击硼,创建一个四面体复合硼与一个负电荷。Baeyer-Villiger一样,有一个o - o键断裂的薄弱迁移σ键:
- 在第一弯箭头,cb,优惠和切断形式
- 在第二曲线箭头,oo的断裂
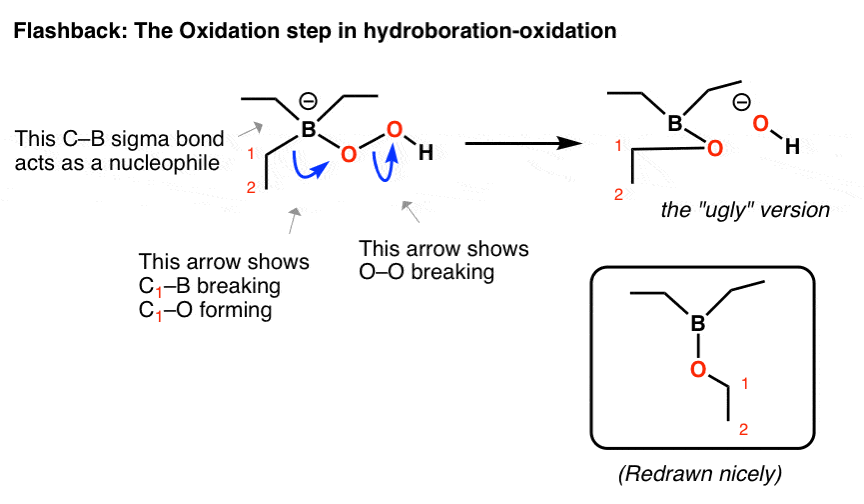
我建议把“丑陋”版本获得正确的连接,然后重新画一下。
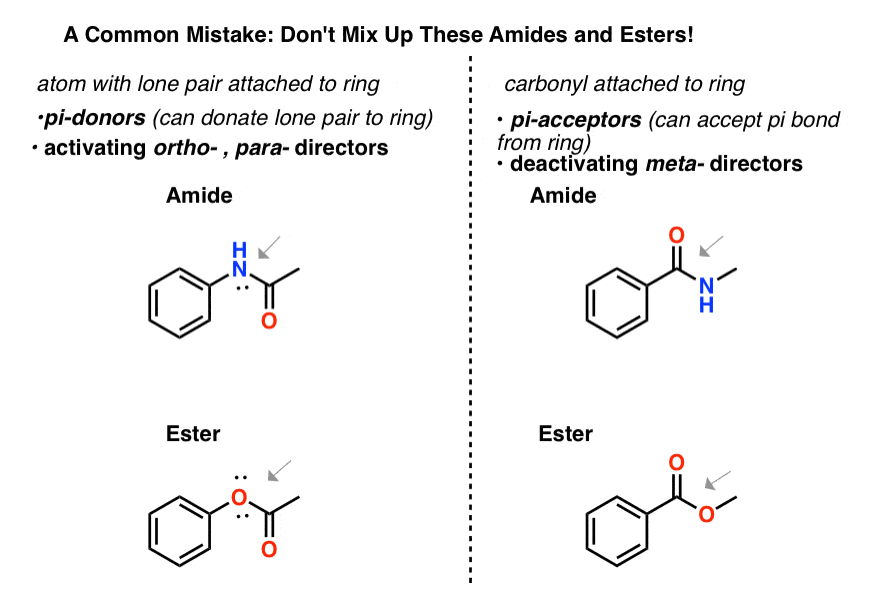
7所示。提醒关于混合的“酰胺”和“酯”
确保你可以告诉酰胺和之间的区别酯类这是昊图公司- - - - - -,帕拉- - - - - -董事、酰胺和酯类这是元——董事。
- 一个原子连接到一个芳环,熊孤对将一个昊图公司- - - - - -,帕拉- - - - - -导演,因为它能够π捐赠到戒指。所以一个酰胺N连接到芳环或一个酯附带一个O的芳环昊图公司- - - - - -,帕拉——董事。
- 一个羰基一个将是一个芳香环元- - - - - -导演,因为它可以接受一个π键的戒指。所以一个酰胺或酯与羰基芳环是一个元导演。
不要让这两个类酰胺和酯类搞混了!
8。摘要:减少硝基组和Baeyer-Villiger氧化
信不信由你,我们几乎覆盖环上的取代反应和反应的芳香族取代基。
那么现在呢?
是时候开始把这些反应在一起序列,试试我们的手合成芳香分子。
所有这些反应在我们的工具包,我们就能完成很多。
在接下来的文章中,第一个在芳香族合成,我们将讨论一些基本的原则。第二个和第三个帖子将涵盖一些合成策略。最后,我们可以花一些时间练习问题。
笔记
注1。。硝基还原的机理不一般了,因为它是loooong(15 +的步骤),时间是有限的,主题不复发的机制在后续的部分课程。这并不是说它并不重要,选择不可避免地需要做的只是,硝基还原必然最终在切割室地板上。这是大致的反应会是什么样子Sn和酸徘徊在弹出图像(链接到图片)
{kind=link}
注2。切断(π)债券形成技术只是一种共振,所以可以或者画一个协调一致的过程。为切断π键的形象参与盘旋((链接到图片)
{kind=link}
好吧,这个东西远远超出基础入门课程的印象,所以随时跳过。不过,问题是:为什么迁徙的奇怪的订单秩序Baeyer-Villiger吗?
三级>二级,芳基>主>甲基
(主要是)在熟悉的碳正离子的稳定性顺序(三级>二级>主>甲基)芳基介于二级和初级除外。现在我们看到很多例子有力地表明,芳基碳正离子比小学更不稳定的碳正离子。给什么?
我们最好的假设是,它实际上并不通过直接的碳碳σ键的迁移,但实际上通过攻击的氧气芳环的pi系统。这个结果在一个非定域化的碳正离子(一个“arenium离子”)高度稳定的共振,迅速分解,恢复最后的途中芳香性酯产品:
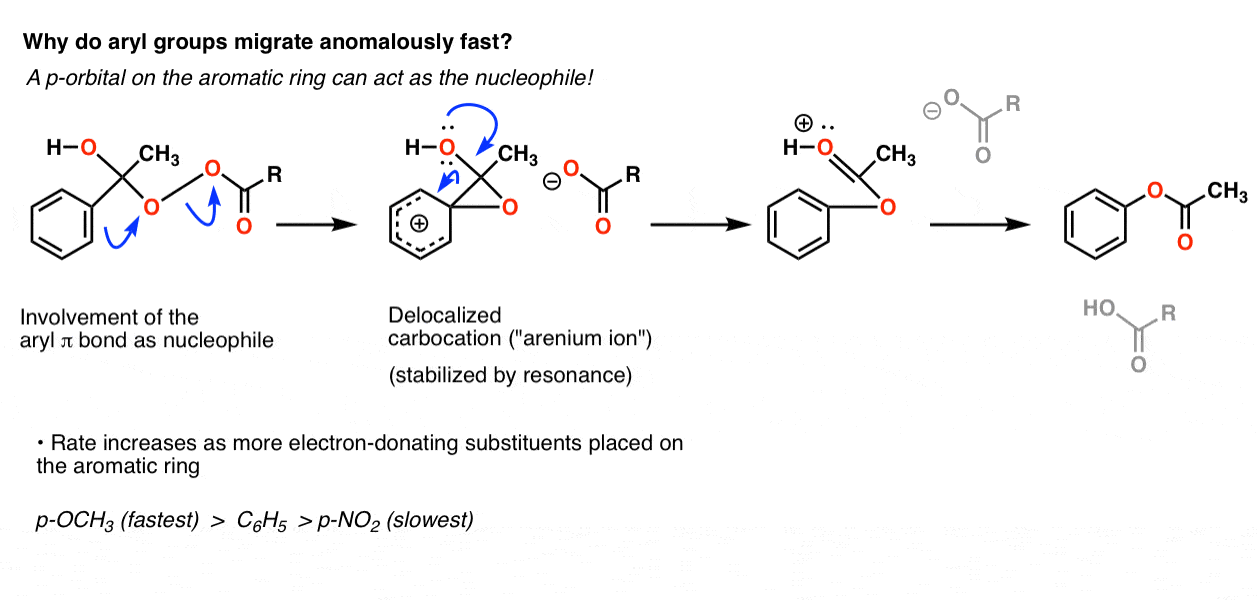
我们希望从这个机制,芳环上的电子基组(比如O-CH3相对于C)的反应速度6H5本身,和吸电子集团(如没有2)延迟率。
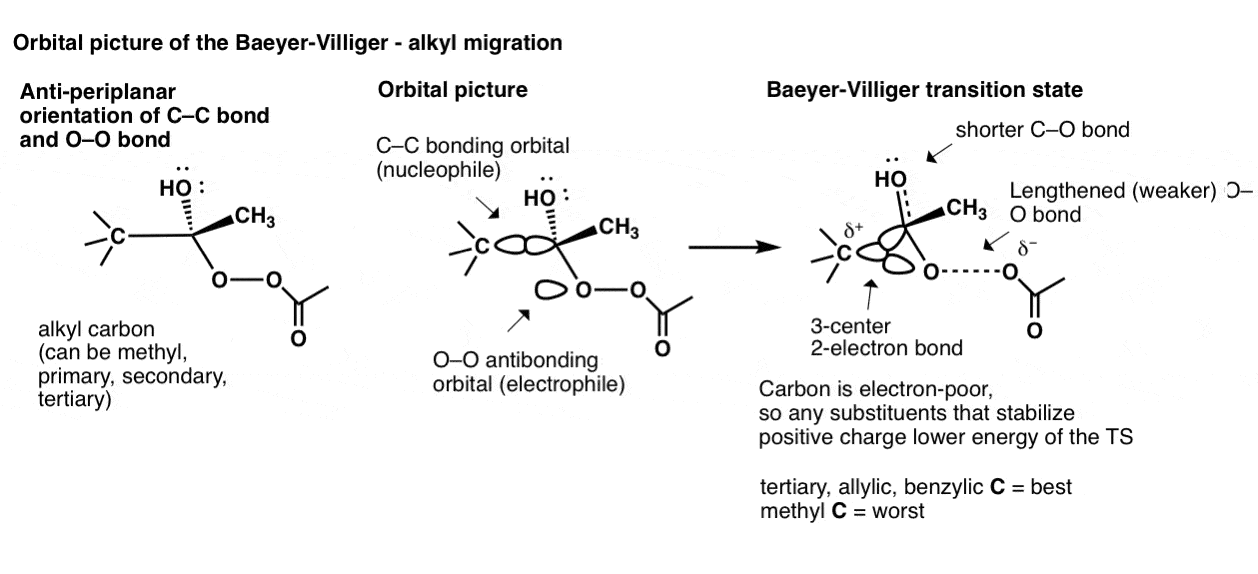
去更深的轨道图我们可以看看烷基Baeyer-Villiger迁移。这始于一个碳碳键对齐反-periplanar安排与oo的债券。碳碳键轨道然后开始重叠与oo的反键轨道,导致一个过渡态,有三个原子共享两个电子!(这就是所谓的,不足为奇的是,“3-center,电子债券”。
这个过渡态的稳定性直接相关的三组烷基碳稳定正电荷。因此,三级(以及烯丙基的和苄基的)碳迁移速度最快,其次是次要的,主要和甲基。
芳基的迁移不仅仅涉及碳碳σ键;上的p轨道碳也可以参与!
所以在这个过渡状态会发生什么p轨道捐赠到oo的反键轨道,导致正电荷的过渡态分散在五个碳的芳环;不3-center、电子债券。
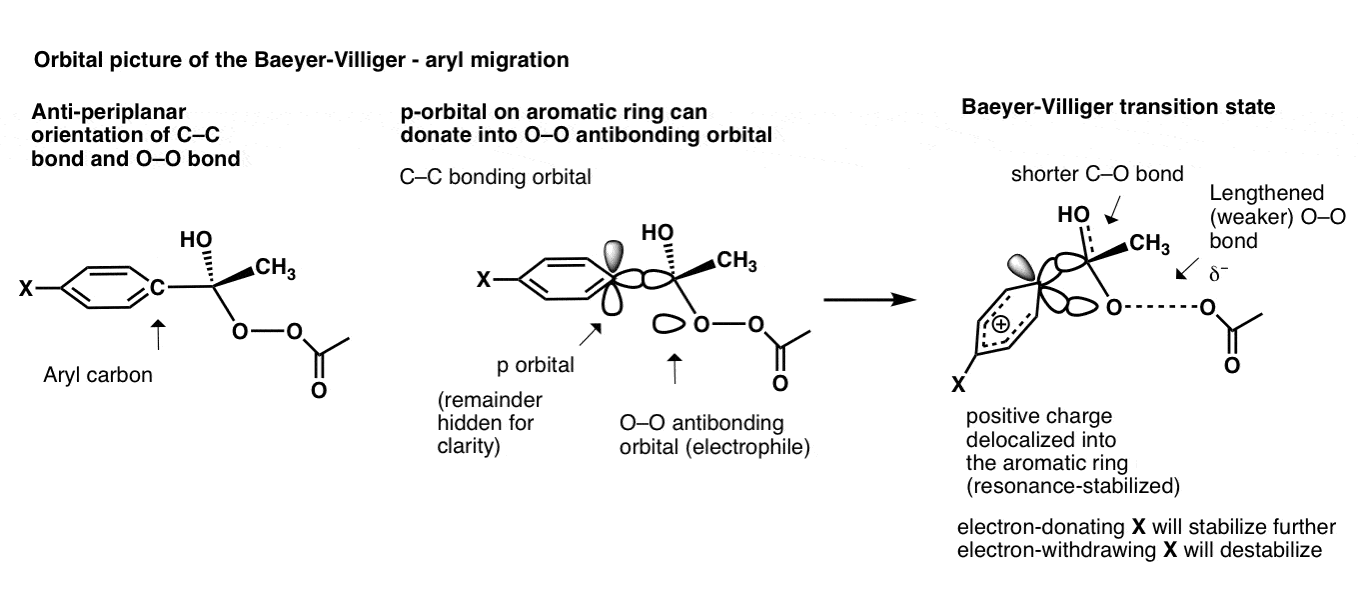
你可以想象,电子捐赠团体“X”如哟3稳定过渡态(增加率)和NO2破坏过渡态等吸电子集团(减缓)。
理论有机化学家亨利Rzepa有有用的博客文章讨论的机制Baeyer-Villiger(尤其是质子转移步骤)在这里。
测试你自己!
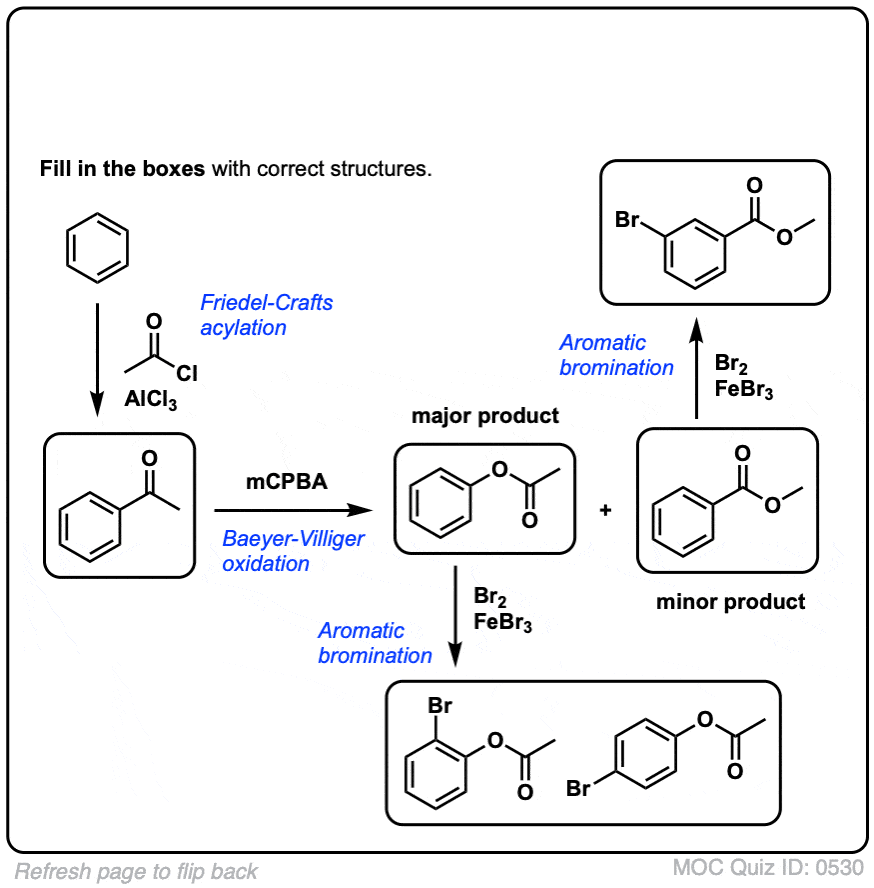
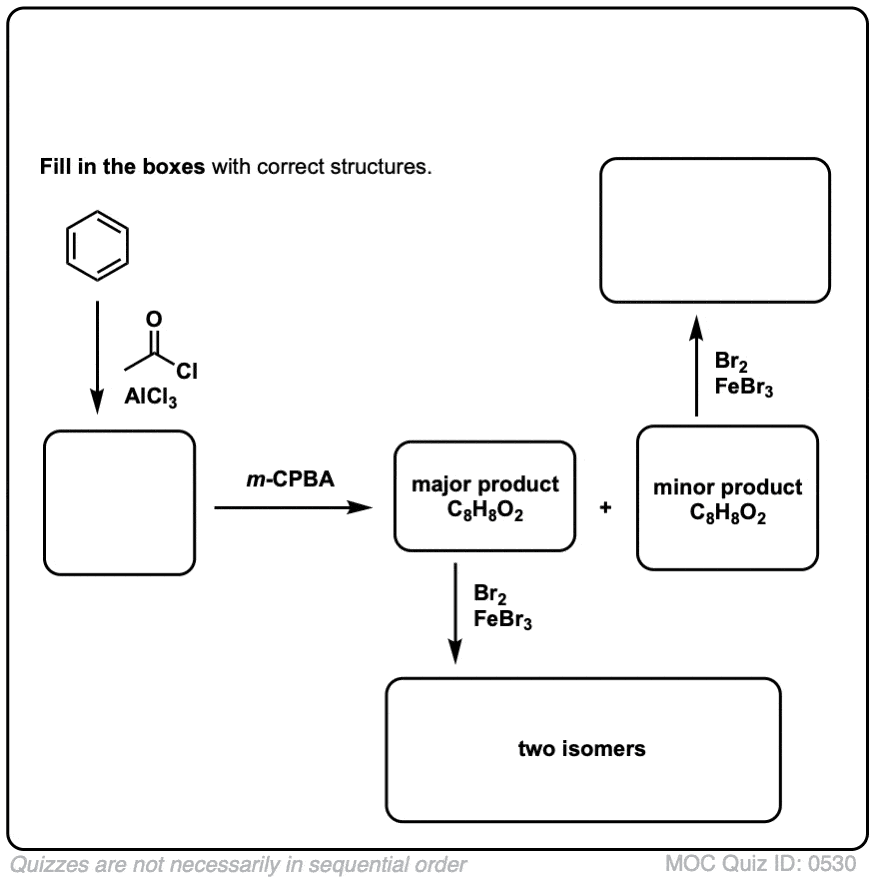 点击翻转
点击翻转
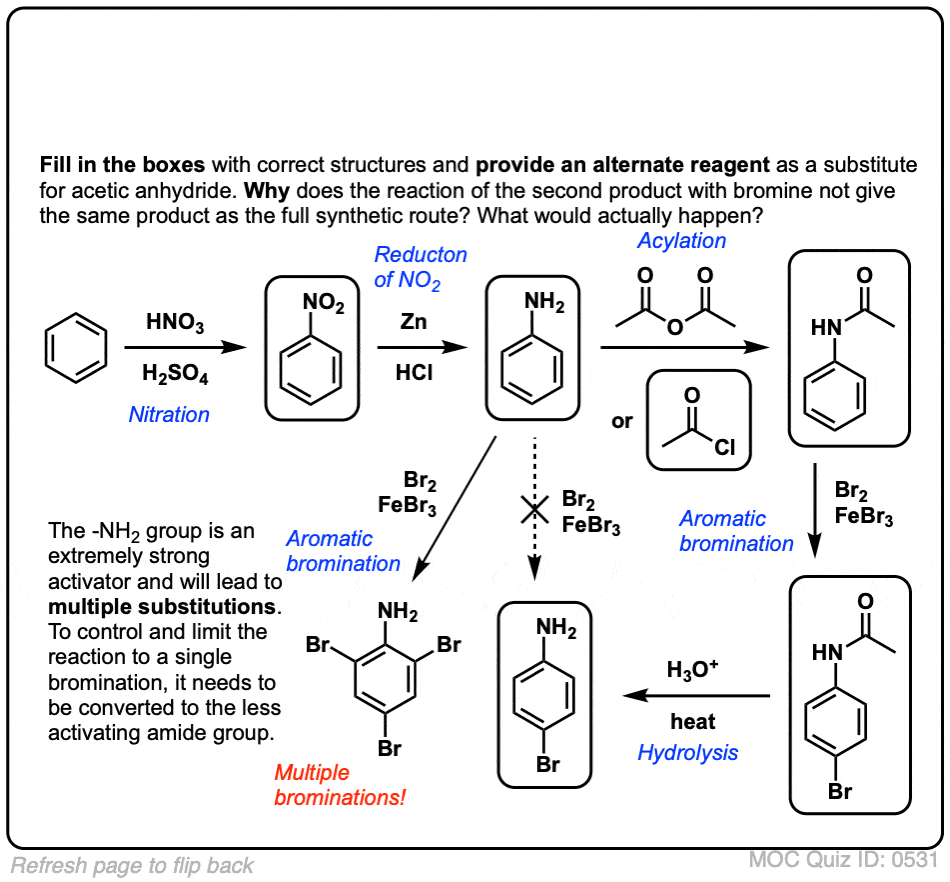
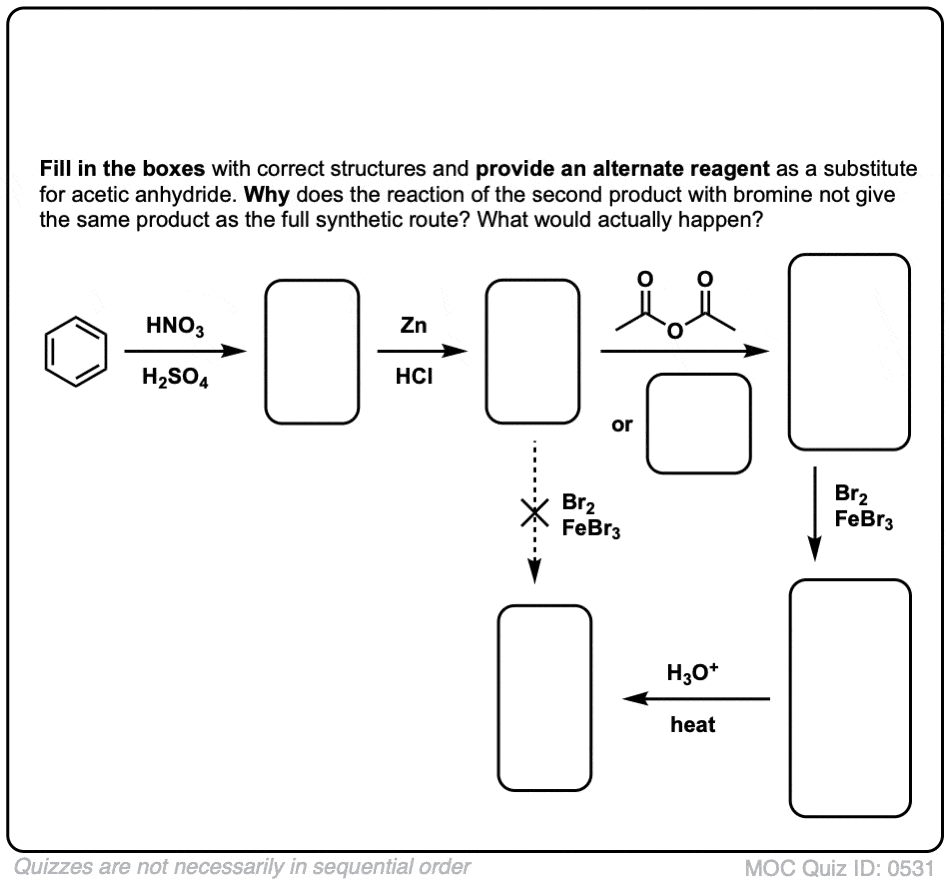 点击翻转
点击翻转
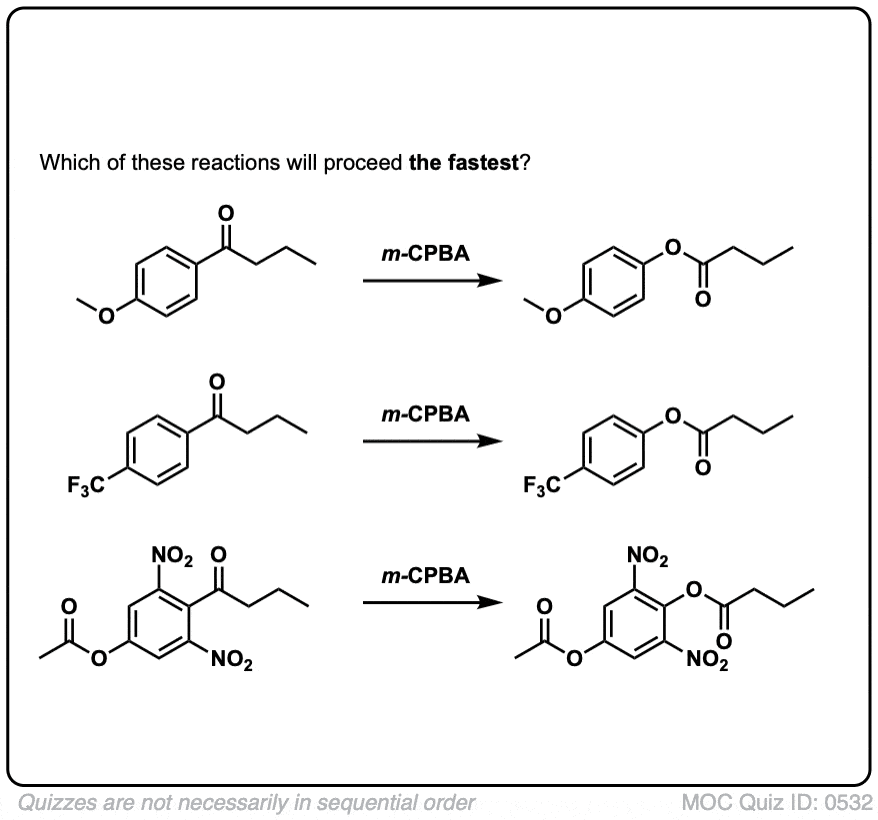 点击翻转
点击翻转
(高级)引用和进一步阅读
Baeyer-Villiger氧化:
- Einwirkung des卡罗'schen化学试剂酮
阿道夫•冯•拜尔,维克多威利格
的误码率。1899年,32(3),3625年
DOI:10.1002 / cber.189903203151
本文由诺贝尔奖得主阿道夫·冯·贝耶尔首先描述了现在被称为Baeyer-Villiger重排,使用过硫酸钠和浓硫酸的混合物(卡罗酸)。 - 23。合成方法.alpha.-methylene可以利用。乙烯酮内酯通过环加
阿尔弗雷德·哈斯内尔哈罗德·w·Pinnick和杰·m·Ansell
《有机化学》杂志上1978年43(9),1774 - 1776
DOI:10.1021 / jo00403a032
本文代表过程Baeyer-Villiger氧化的实验部分。 - 100年的Baeyer-Villiger氧化反应
迈克尔还建议、莫尼耶伯纳德
欧元j . Org。化学。1999年,4,737年
DOI:1002 / (SICI) 1099 - 0690 (199904) 1999:4 < 737:: AID-EJOC737 > 3.0.CO; 2 b
Baeyer-Villiger氧化的审查包括一个详细的历史视角的发展和历史的反应。 - Baeyer-Villiger氧化酮和醛
Krow, g R。
Org。反应。1993年,251年
DOI:10.1002/0471264180. or043.03
这么长时间,详细的审查包括机制的深入讨论,底物范围,限制,和实验Baeyer-Villiger氧化过程。 - 威利格拜耳−反应:新发展对环保手续
- j。十边缘,我。w·c·e·阿伦兹,r·a·谢尔登
化学评论2004年104年(9),4105 - 4124
DOI:10.1021 / cr030011l
综述了现代视角Baeyer-Villiger氧化和描述程序使用更环保的氧化剂(如O2)。 - Baeyer-Villiger氧化与过氧化氢催化芳香醛和酮的硒化合物。一个方便的酚类化合物的制备方法
Ludwik syp
合成1989年,3,167 - 172
DOI:1055 / s - 1989 - 27183
芳基/ H的迁移很少发生,出现在一些例子。 - 实验支持主要的立体效果管理拜耳−威利格氧化和Criegee重排
理查德·m·古德曼和Yoshito岸
美国化学学会杂志》上1998年,120年(36),9392 - 9393
DOI:10.1021 / ja982188g
一个简短的交流一些基质用于b对氧化产生产品证明立体电子效应迁移能力的原因。Yoshito岸教授被认为是哲学继承了传奇化学家r·b·伍德沃德,他继承了伍德沃德教授的研究小组伍德沃德去世后,继续挑战总合成的伍德沃德的遗产,并在海洋天然产物的合成水螅毒素,被认为是迄今为止最具挑战性的合成。岸教授也有他的名字与反应相关联,Nozaki-Hiyama-Kishi反应的机理,发现了,而偶然发现的。还原芳香硝基组:
可用多种方法降低,可以通过加氢或使用金属在酸。 - 2-AMINO-p-CYMENE
f·h·艾伦和詹姆斯VanAllan
Org。Synth。1942年,22,9
DOI:10.15227 / orgsyn.022.0009
减少一个早期的例子有机合成,可靠的来源和独立进行测试有机合成实验室程序。这个使用加氢兰尼镍减少硝基。 - 减少氢化铝锂的有机化合物。三世。卤化物、醌类、杂氮的化合物
罗伯特·f·Nystrom和韦尔登·g·布朗
美国化学学会杂志》上1948年,70年(11),3738 - 3740
DOI:1021 / ja01191a057
LiAlH4还可以用于降低芳香族硝基组,但这将给偶氮苯。这似乎是一个非常方便的偶氮苯的合成方法,如p-nitrobromobenzene——“中产品从减少p-nitrobromobenzene(室温),4,4′-dibromoazobenzene,不溶于水,略溶于醚以晶体的形式,在稀硫酸。获得非常纯形式仅仅通过过滤,用热水洗涤。” - 的选择性还原芳香硝基化合物在非酸性氯化亚锡和非水介质
d·贝拉米Ou
春节。列托人。1984年,25(8),839 - 842
DOI:10.1016 / s0040 - 4039 (01) 80041 - 1
SnCl2在乙醇可以用作pH-neutral,硝基还原非水系统。 - 还原芳香硝基化合物的二级醇使用铑配合物作为催化剂
f . Liou和c·h·程
《有机化学》杂志上1982年,47(15),3018 - 3021
DOI:10.1021 / jo00136a046
数以百计的变体中已知的这个反应,Rh复合物可以作为同类催化剂的加氢还原硝基的转移。
谢谢你的帮助给了如此详细的东西
詹姆斯,一个评价。在测试的第二个问题,你应该提到,除了控制稀稀拉拉/ para烷基化形成苯胺还meta-alkyl使用免费的(根据缺点胺在酸性条件下)。
如果我没有错误的迁徙能力是H(最快)>苯组>三级>二级>主(慢)
不是吗?
从3月/史密斯,先进的有机化学第五。”不对称酮的大致顺序迁移叔烷基>二级主烷基烷基和芳基> >甲基。
之前我会把H叔烷基,尽管显然芳基/ H发生迁移(很少)。https://www.thieme - connect.com/products/ejournals/abstract/10.1055/s - 1989 - 27183