芳香分子的反应
东亚峰会(3)——傅克酰化和傅克烷基化反应
最后更新:2022年9月28日|
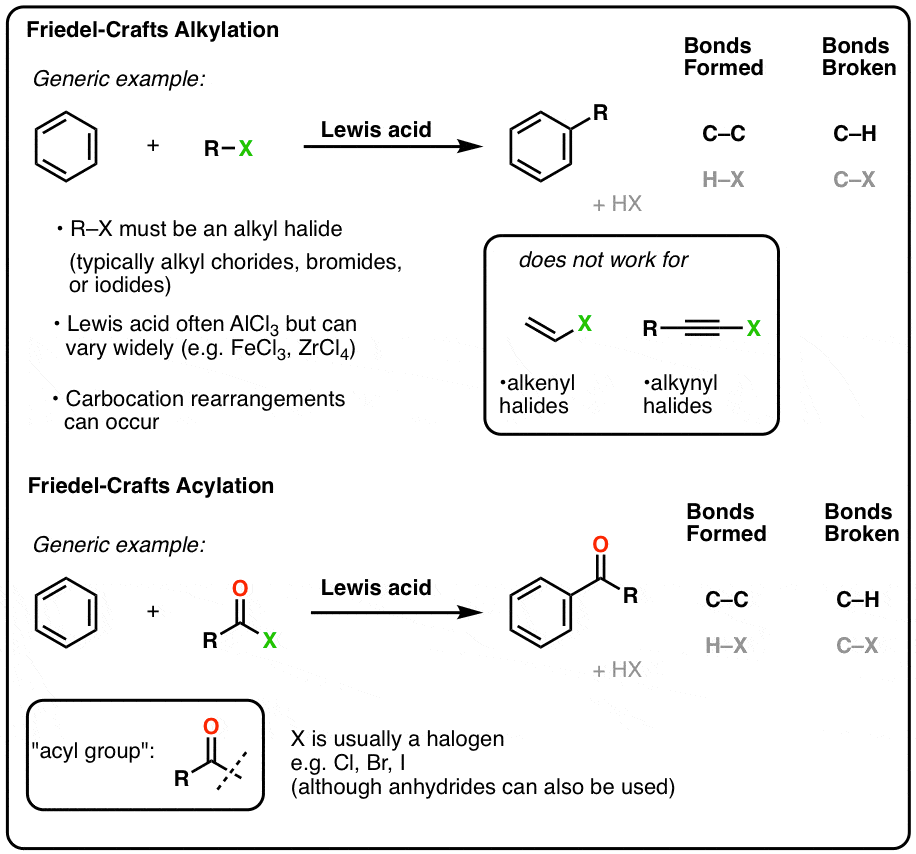
弗里德工艺品烷基化和酰化
- 处理时将形成碳碳键的芳香环烷基或酰卤化物在存在强烈的路易斯酸(例如AlCl3)。这些被称为弗里德工艺品反应的例子亲电芳香取代反应。
- 路易斯酸坐标上孤对卤素、卤素更好离去基团。
- 在一个傅克烷基化烷基卤化物与路易斯酸治疗导致碳正离子亲电试剂(或一个物种非常类似于碳正离子)由芳环,然后攻击。
- 碳正离子重排可能发生如果他们的结果在一个更稳定的碳正离子!
- 傅克酰化与执行酰卤化物和路易斯酸。没有重组是观察到的。
表的内容
- 快速回顾亲电芳香取代:起碳碳键形成反应!
- 芳香环的傅克烷基化
- 路易斯酸的傅克烷基化作用是激活烷基卤化物
- 碳碳键形成的关键一步是攻击的芳环上碳正离子(或carbocation-like)亲电试剂
- 小心!碳正离子重排可能发生傅克烷基化反应
- 如果一个氢化物转移或烷基转移将导致一个更稳定的碳正离子,假定它会发生
- 傅克烷基化的限制
- 傅克酰化
- 傅克酰化反应的机理
- 没有重组发生傅克酰化
- 傅克酰化的限制
- 摘要:傅克烷基化和酰化
- 笔记
- 测试你自己!
- (高级)引用和进一步阅读
1。快速回顾亲电芳香取代:起碳碳键形成反应!
这是第三次在一系列的三个职位的关键亲电芳香取代(EAS)在介绍有机化学反应。
综上所述,到目前为止,我们已经学了反应形成carbon-halogen,碳氮和碳硫键。
到目前为止什么重要的类债券失踪的?
碳碳键的形成反应!(注1]
在这篇文章中,我们将介绍两个重要的碳碳bond-forming亲电芳香取代反应熊发现者的名字,查尔斯·弗里德和詹姆斯·工艺:傅克烷基化和傅克酰化。
我们还将看到,这些反应遵循熟悉三步模式在以前的亲电芳香取代反应,即:
2。芳香环的傅克烷基化
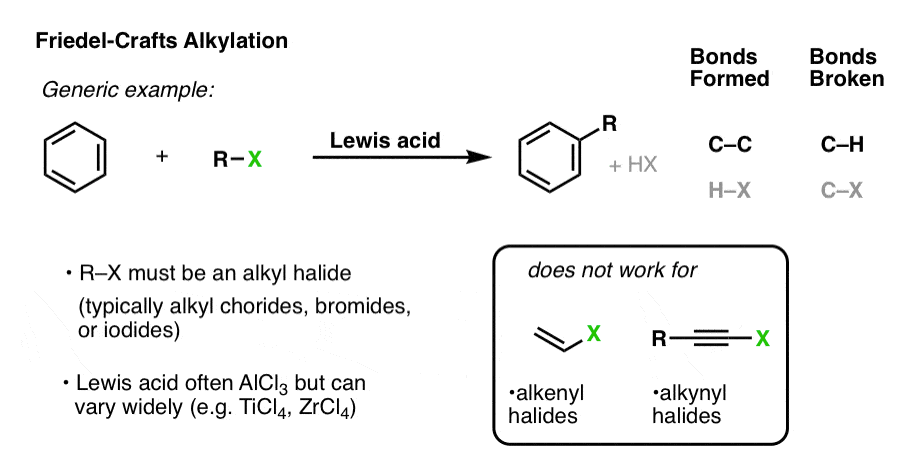
当一个烷基卤化物与路易斯酸在治疗一个芳环的存在,烷基组可以被添加到环(形成碳碳)和碳氢键的损失。这亲电芳香取代反应被称为傅克烷基化的反应。
一般来说,没有反应发生在路易斯酸的缺失。路易斯酸的一个常见的选择是氯化铝,间变性大细胞淋巴瘤引起3,但很多人可能被使用,如FeCl3等等。
烷基卤化物(通常为氯化物、溴化物和碘化)必须使用,反应不完全烯基和炔基卤化物。
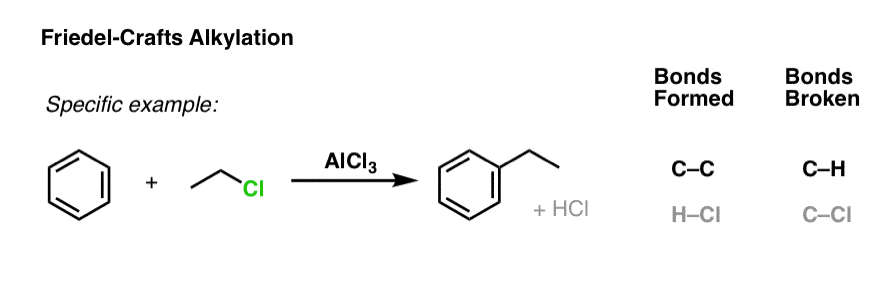
这是一个特殊的例子使用氯乙烷:
3所示。路易斯酸的傅克烷基化作用是激活烷基卤化物
如果没有反应发生在缺乏路易斯酸,然后这里的路易斯酸的作用是什么?
就像我们在前两个帖子看到亲电芳香取代反应,路易斯酸“激活”亲电试剂通过协调离去基团,使其成为较弱的基础,和一个更好的离去基团(AlCl4- - - - - -是一个较弱的比Cl基地吗- - - - - -)。最终的结果是,协调的路易斯酸亲电试剂使这个物种更好的亲电试剂。
例如,使用异丙基氯化(下图),第一步是协调AlCl的协调3氯原子。这削弱了C-Cl键,结果Cl可以离开(AlCl4- - - - - -)给一个仲碳(一个更好的亲电试剂比异丙基氯本身)。
- 二、三级卤化物,充分游离碳正离子可以发生。
- 主要的(甲基)烷基卤化物,亲电试剂很可能不是“免费”碳正离子,但“carbocation-like”物种C-Cl债券是大大削弱/延长。
- 我们简要地提到的,没有反应发生烯基或炔基卤化物,很大程度上是因为这些物种是如此不稳定的碳正离子和难以生成。
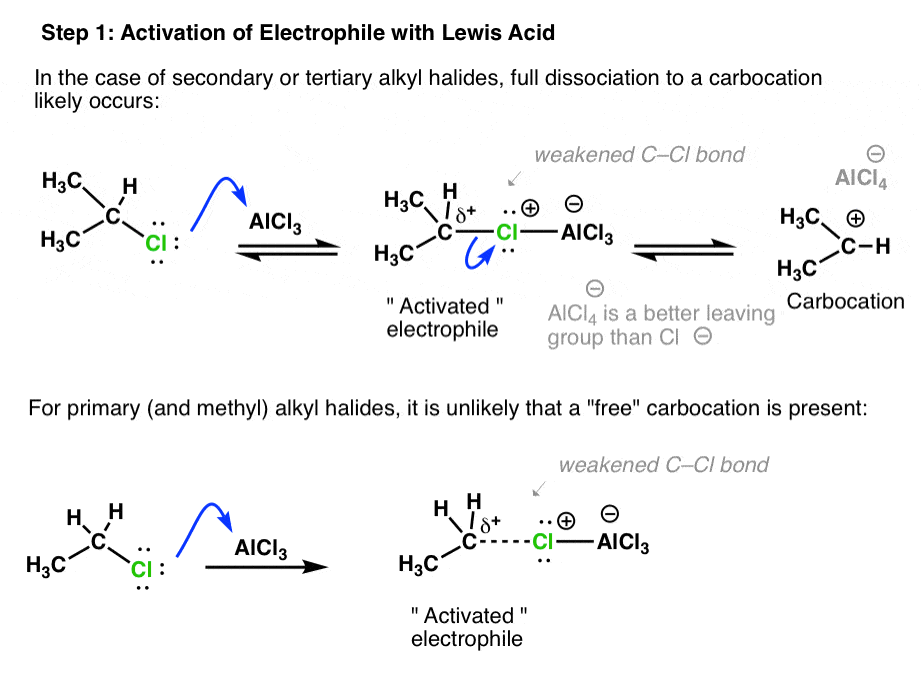
注意:尽管我们显示碳正离子亲电试剂傅克反应中生成的烷基卤化物和路易斯酸,还有其他的方法来生成碳正离子(如通过烯烃的质子化作用,见下文]。我们一般定义傅克烷基化是一个芳环碳正离子的反应(或carbocation-like)的中间体。看到注2为例。
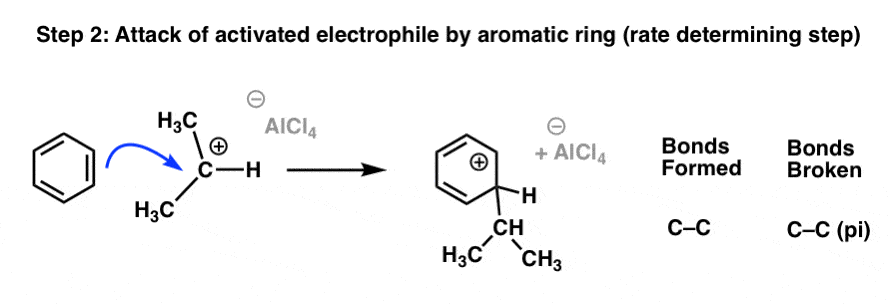
4所示。碳碳键形成的关键一步是攻击的芳环碳正离子(或carbocation-like)亲电试剂
一旦亲电试剂已经激活,下一步的傅克反应激活的攻击亲电试剂芳环。这也是速率决定步骤,因为它会破坏环的芳香性(及其~ 36千卡每摩尔的共振能量)。
在这一步中碳碳(pi)债券的芳环断裂,形成新的碳碳σ键,导致碳正离子中间体:
最后一步是去质子化的碳氢键弱碱(例如Cl- - - - - -)恢复环的芳香性:
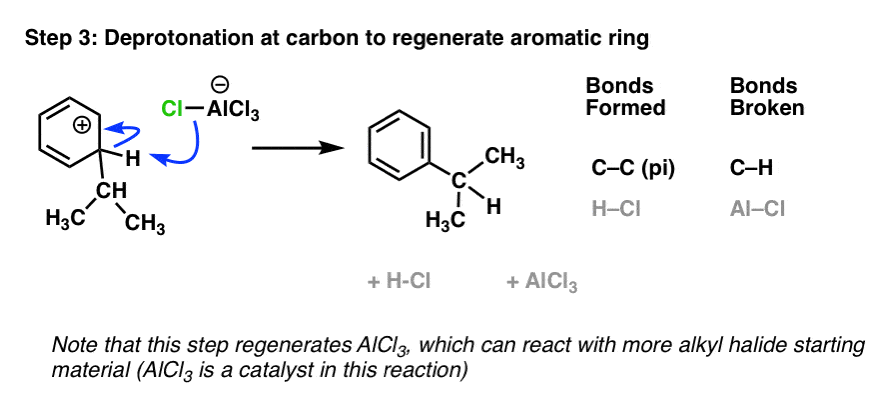
(另一种方法来描述这个反应是游离氯的弯曲的箭头- - - - - -间变性大细胞淋巴瘤引起的4- - - - - -然后使用它作为基地。要么这样,同样的事情)。
请注意,AlCl3再生,允许它再次被使用在步骤1中与另一个的吗烷基卤化物。因此,间变性大细胞淋巴瘤引起3可以作为催化剂在这个反应中,因为它增加了反应速率,但不使用它。
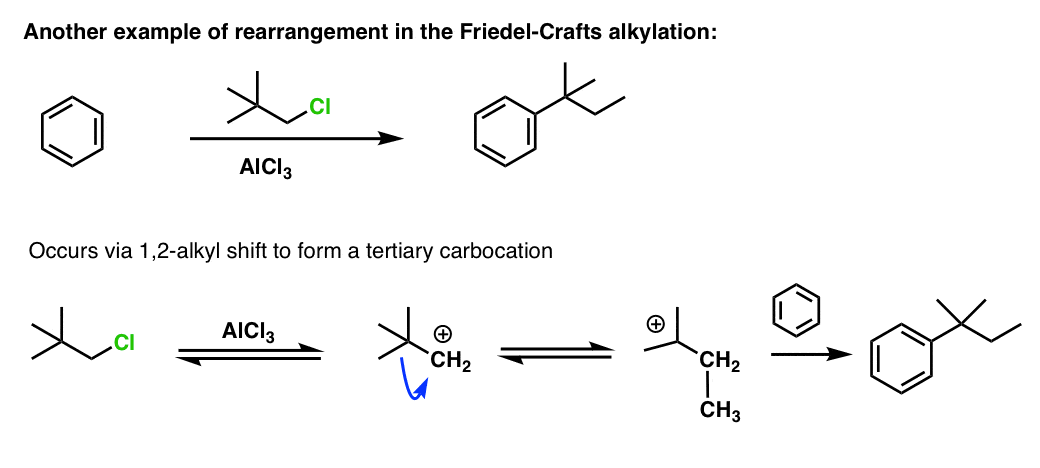
5。当心!碳正离子重排可能发生傅克烷基化反应
许多大学科学课程是教在单位,你在一个模块学习几乎零重叠与你学习在另一个。
不用说,有机化学并不是这样的。你可能已经经历了概念学习情况组织1回荡回到Org 2章节以意想不到的方式。嗯,准备另一个有趣的例子。
我们上面显示乙基氯对苯和间变性大细胞淋巴瘤引起的反应3提供乙苯的傅克烷基化。
扩展这个反应的氯化乙基丙基氯应该相应地给丙基苯。
对吧?
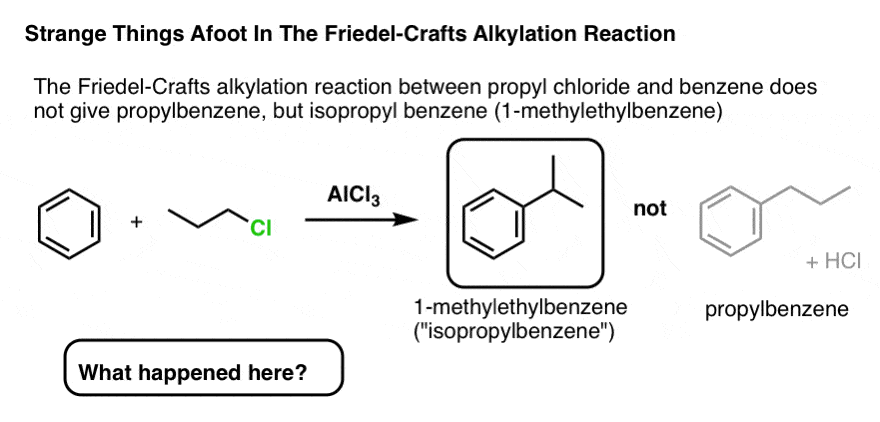
…。茴香素?
这是怎么发生的?
快速的记忆,决定。还记得这心爱的反应从Org 1吗?
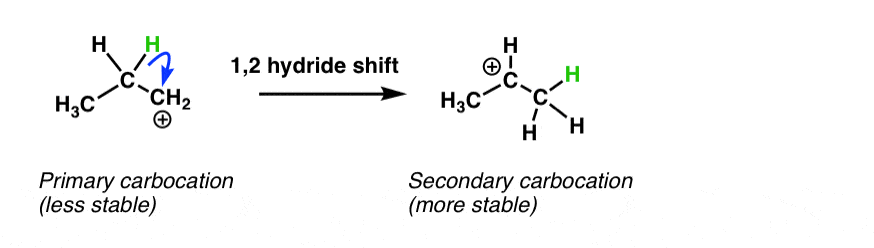
啊,氢化物的转变。碳正离子可以通过氢化和重新排列烷基转移这样一个不太稳定的碳正离子转化为一个更稳定的碳正离子。
傅克反应,我们已经见过一种路易斯酸的协调烷基卤化物导致碳正离子(或主要的烷基卤化物,至少是一种“carbocation-like”),然后被芳环的速率决定步骤。
6。如果一个氢化物转移或烷基转移将导致一个更稳定的碳正离子,它会发生
这里正在发生的事情是没有不同:如果碳正离子可以重新排列一个更稳定的碳正离子通过氢化或烷基转变,它会这样做。
有机化学2:第一学期的课程概念回到咬你的屁股。™
这是在丙基氯的情况下会发生什么。
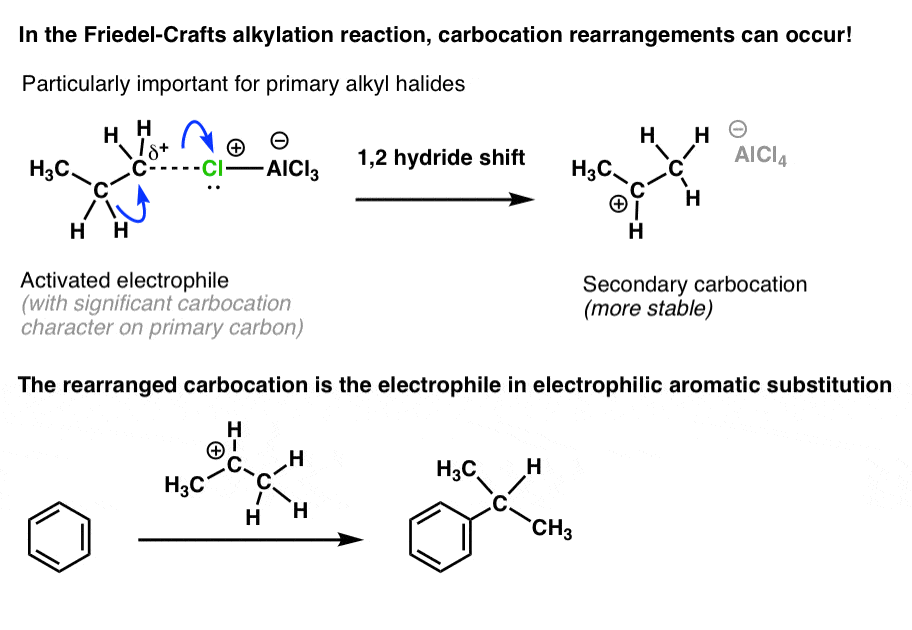
从C氢化物的转变2C1导致二次碳正离子,然后芳环的攻击。
底线的傅克烷基化反应:
在FC烷基化重排的另一个例子包括在脚注只是为了好玩。(注3]
7所示。Freidel-Crafts烷基化的限制
最后注意的傅克烷基化:几个缺点。
- 首先,正如我们所看到的,碳正离子重排可能发生的。(有间接绕过这个问题的方法,下面我们将暗示(跳过到下]。
- 第二,傅克烷基化可能不会奏效electron-poor芳香环,尤其强劲取代基如CF才会安静下来3,没有2,所以3H,等等。卤素是好的。
- 第三,这是比任何其他更实际的问题,所以经常被忽略——FC烷基化的产品通常是一个更好的亲核试剂起始物料。(回想一下,烷基组激活)。结果可以有点像饼干怪兽在芯片喂! !工厂——它不能停在一个,导致多个烷基化。
8。傅克酰化
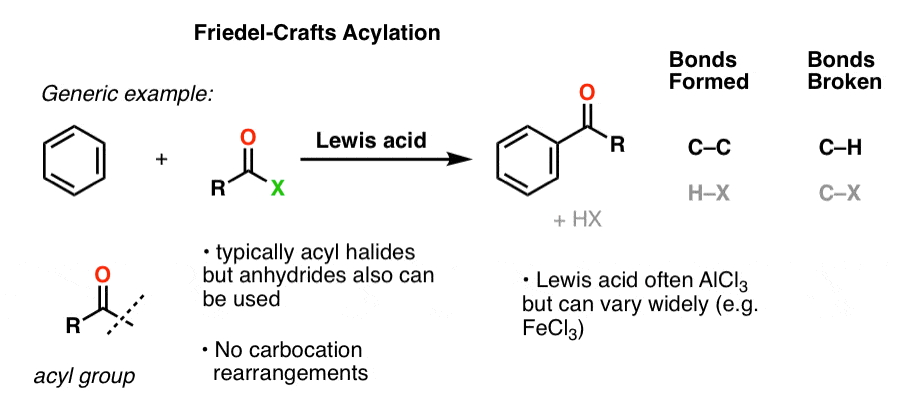
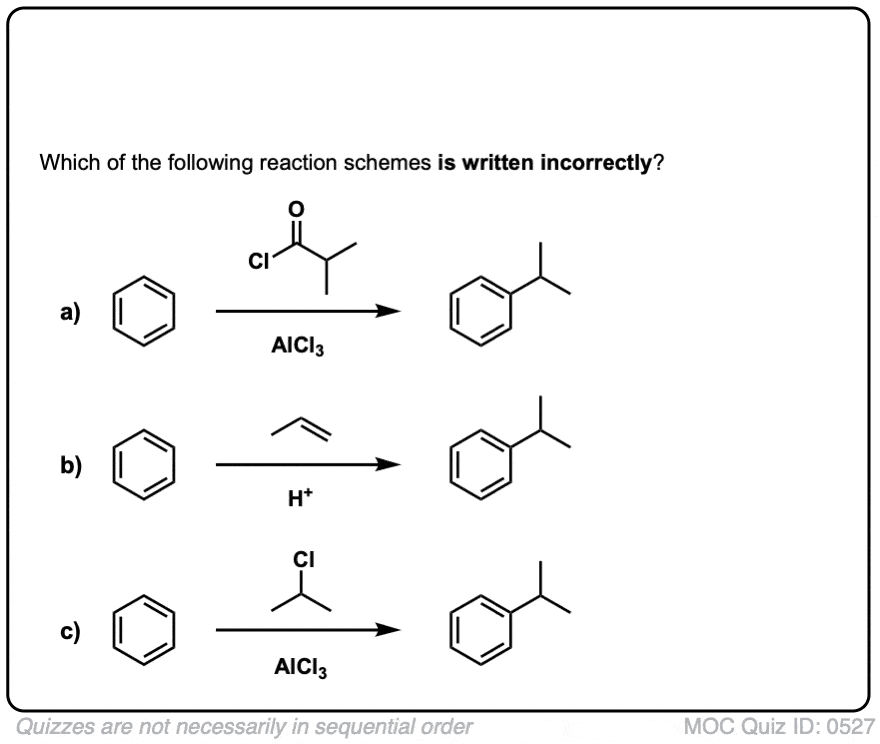
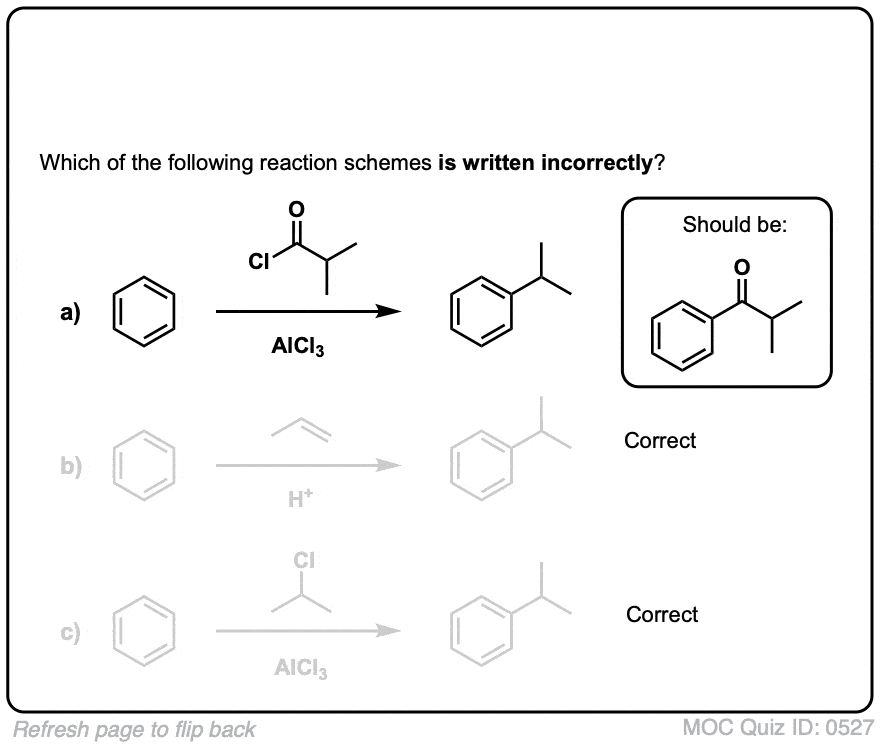
一个与傅克烷基化过程,调用傅克酰化发现了弗里德和工艺大约在同一时间(1877)。如果路易斯酸添加到一个酰卤在一个芳环的存在,一个亲电芳香取代反应可能发生,酰基集团增加了芳环(HX的损失)。
与足球烷基化,具体傅克酰化的路易斯酸可以有所不同。氯化铝(AlCl3)是经常使用,但FeCl3和其他路易斯酸也会做这项工作。
这里有一个通用傅克酰化的例子:
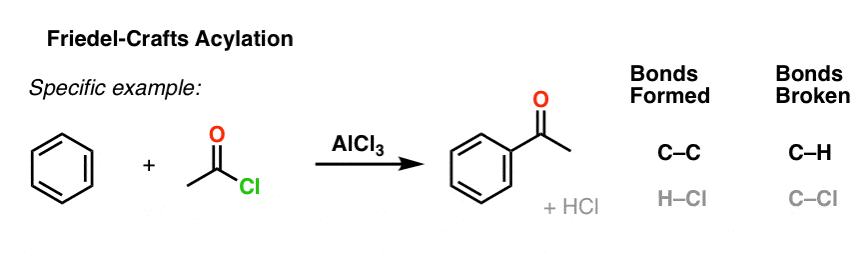
一个具体的例子之间的反应是由铝氯乙酰氯和苯催化:
9。傅克酰化反应的机理
所以傅克酰化反应是如何工作的呢?
与FC烷基化,第一步是激活的亲电试剂。路易斯酸坐标卤素、卤素(AlCl的离开4- - - - - -)结果相当稳定,共轭碳正离子上知道“acylium离子”。
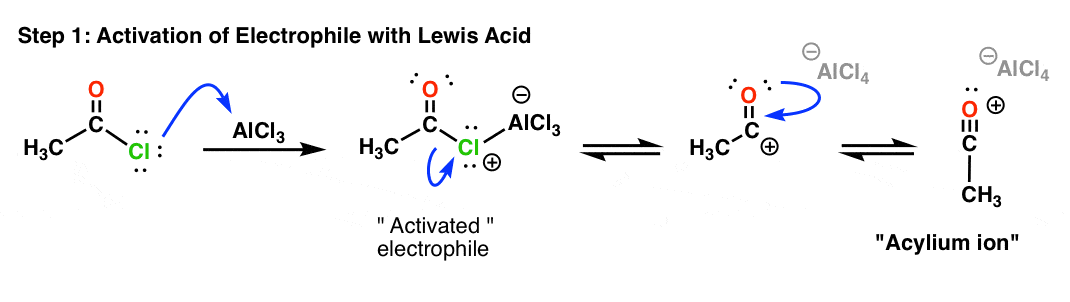
acylium离子是活跃的亲电试剂在傅克酰化反应。一旦形成,acylium离子由芳环的攻击:
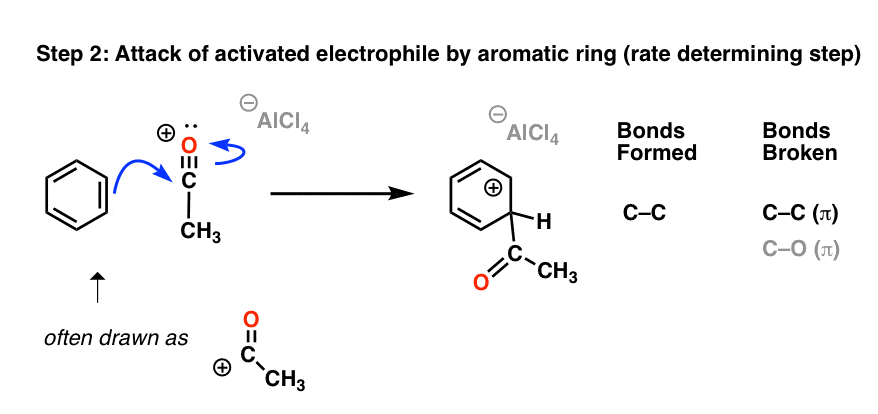
与傅克烷基化,最后一步是去质子化在芳环碳再生。
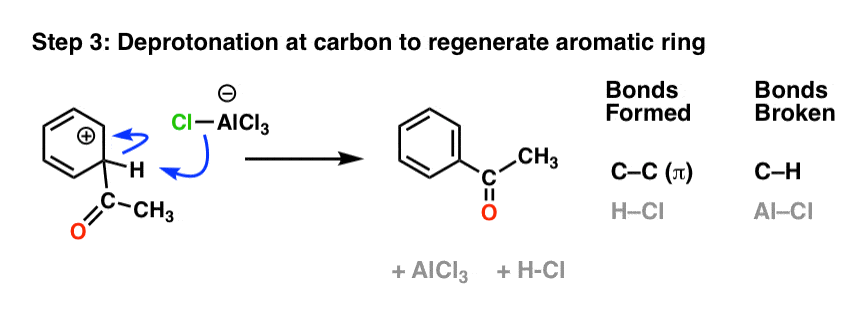
10。没有重组发生傅克酰化
与傅克烷基化,没有发生重排傅克酰化。
这开辟了一个“解决方案”使用傅克酰化得到产品,否则很难获得通过傅克烷基化碳正离子重排。(我们将在以后的文章中详细讨论这个,但是我们给一个口味)。
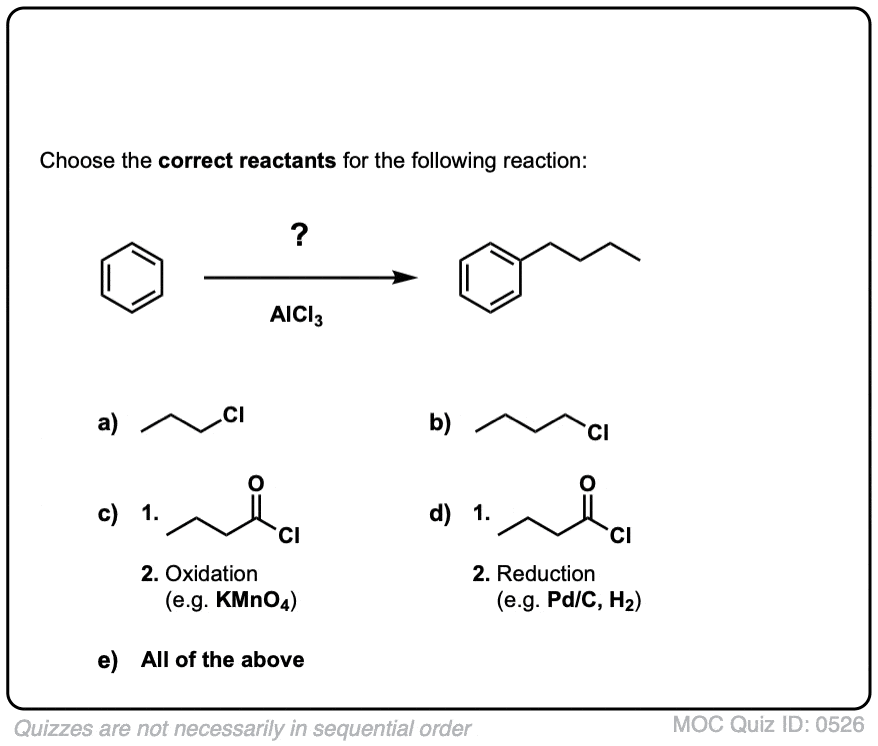
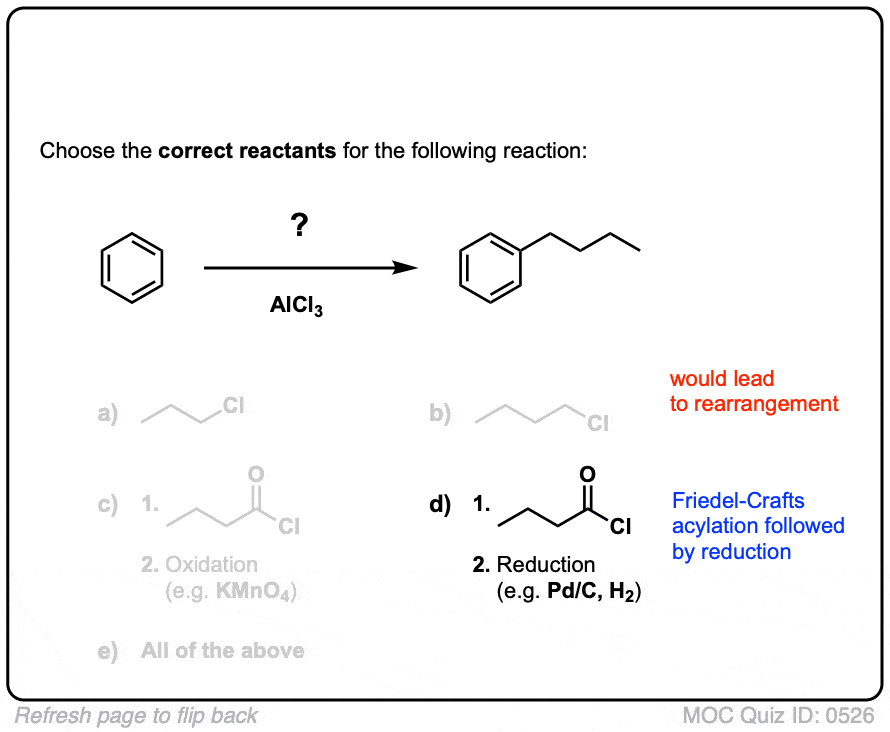
例如,让我们看一下如何使用这个生产丙基苯,我们看到不能由苯与AlCl的傅克烷基化反应3和1-propylchloride。
这里的第一步是进行傅克酰化反应在苯和propionylchloride之间,也许AlCl的催化3。这给了我们乙苯基酮。
下一步是执行的减少酮烷烃,(我们很快就会看到)可以以不同的方式执行。这给了我们1-propylbenzene。
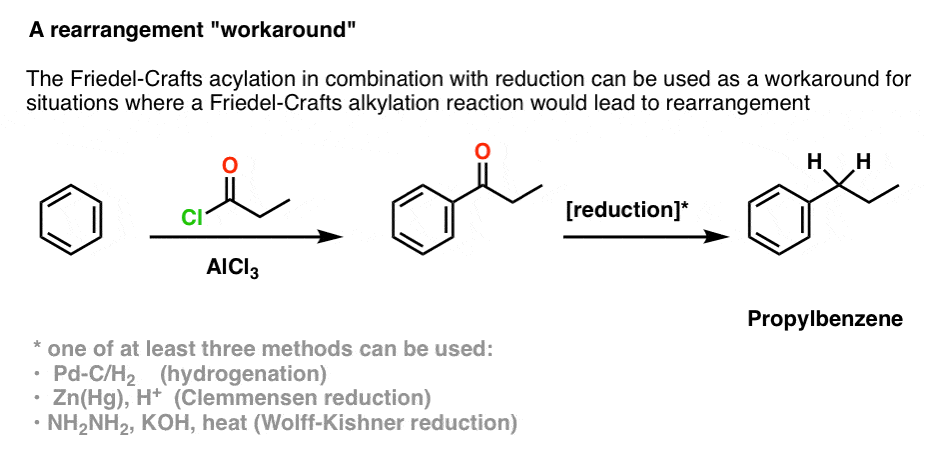
[评论者维克多,从化学帮助中心,帮助指出,有一个第四的方法——转换酮thioketal,然后减少下来的烷烃兰尼镍。]
11。傅克酰化的限制
- 类似于烷基化、傅克酰化往往失败在芳香环与强烈的去活化组织如硝基、CF3,磺酰等等。然而,卤代芳烃仍然工作。
- 把这“可能不需要知道类别”,但傅克酰化催化剂营业额不是很好。在“现实生活”中,间变性大细胞淋巴瘤引起的化学计量量3通常是需要自AlCl吗3坐标强烈酮产品。
12。摘要:傅克烷基化和酰化
因为他们形成碳碳键,傅克烷基化和酰化反应尤为重要亲电芳香取代反应。与溴化、氯化、硝化和sulfonylation他们六个核心亲电芳香取代反应。
在我们完成治疗亲电芳香取代,值得进入细节的一个方面经常给学生头痛的傅克反应;的分子内的版本。
在下一篇文章:分子内的傅克反应
笔记
注1。加分如果你说“碳氧”类型的债券形成的我们还没有看到在东亚峰会。直接亲电氧化苯环是很难在实验室做的。对于我们的目的,形成切断一个芳环通常做的间接,靠的是手段而不是一个直接的东亚峰会。两种方法我们将探讨在适当的时候Baeyer-Villiger氧化和某些重氮盐的反应。
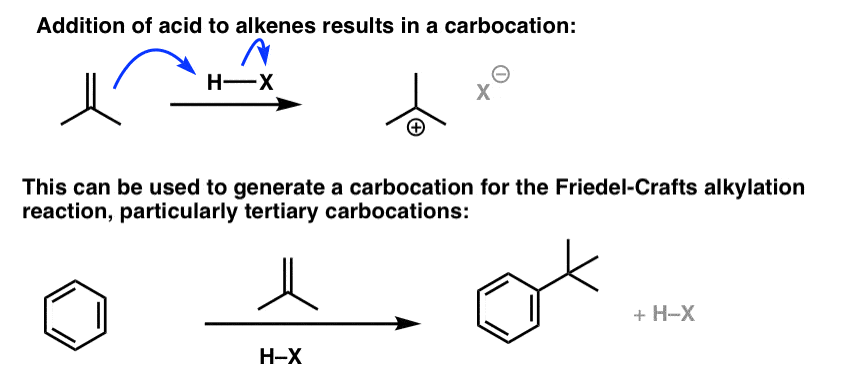
注2。的另一种方式进行傅克烷基化是生成的碳正离子通过质子化作用烯烃。这效果最好,当一个相当稳定的碳正离子是生成的,如t丁基碳正离子通过质子化作用生成异丁烯。
注3。奖金的例子烷基转变。
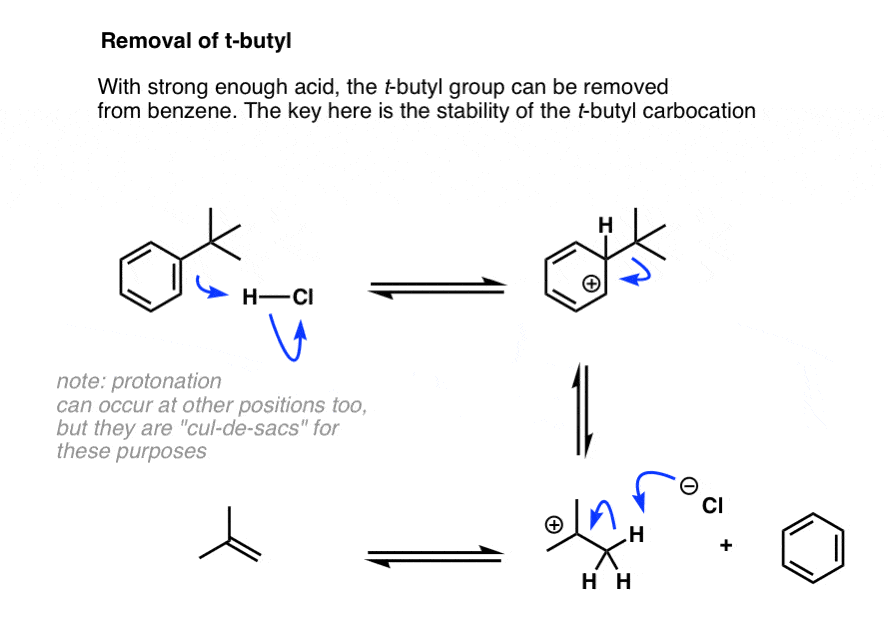
注意4。最后一篇文章里,我们得知磺酰组与强酸可以删除,我提到另一组,可以删除,覆盖下一篇文章(即这篇文章)。这个群体是t丁,可以迫使条件下,与强酸。这工作,因为t丁基碳正离子的相对稳定和反向傅克烷基化是可行的。
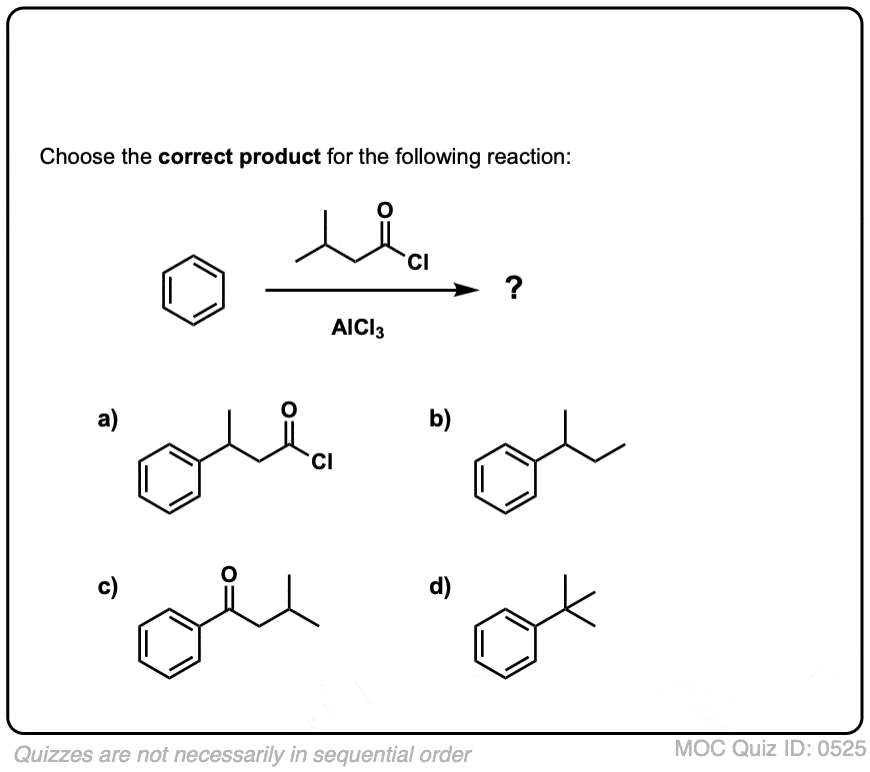
测试你自己!
 点击翻转
点击翻转
 点击翻转
点击翻转
 点击翻转
点击翻转
 点击翻转
点击翻转

 点击翻转
点击翻转
(高级)引用和进一步阅读
傅克烷基化:
- 杰杰克李在他的书中描述名字的反应:
“发现的傅克反应是意外的结果和敏锐的观察。1877年,弗里德和工艺都是在查尔斯·a·维尔茨的实验室工作。为了准备戊碘,他们对待戊基氯碘化铝和使用苯作为溶剂。碘化戊,他们最终与戊苯!不像其他那些可能简单地丢弃的反应,他们彻底调查刘易斯催化烷基化和酰化和发表了50多篇论文和专利的傅克反应,已成为一个最有用的有机反应。”
DOI:10.1007 / 978 - 3 - 642 - 01053 - 8 - _101 - 一个新式方法兴业银行德综合d 'hydrocarbures d 'acetones等等。
弗里德和j . m .工艺品
点数。撕裂。1877年84年,1392 - 1395
经典的,原来在法国由弗里德和工艺品烷基化芳烃(苯在这种情况下)烷基氯化物与AlCl3。 - 重组的傅克烷基化苯
亨利·吉尔曼和r . n .餐
《有机化学》杂志上1943年,08年(2),126 - 146
DOI:10.1021 / jo01190a003
本文通过传说中的美国化学家教授亨利·吉尔曼(爱荷华州)仔细研究苯的烷基化长链烷基卤化物(C10和上图)。这是一个巨大的努力,特别是考虑到这是前现代色谱和光谱技术(如GC或核磁共振),这将使混合物的分析和表征这些化合物容易得多。乔治•a . Olah获得了1994年诺贝尔化学奖,是著名的为他在超强酸和傅克反应化学工作。下面是选择他的论文相关的傅克烷基化: - 芳香取代反应。VI。中间复合物和傅克反应的反应机理烷基化和酰化
g . a . Olah和s·j·库恩
美国化学学会杂志》上1958年,80年(24),6541 - 6545
DOI:10.1021 / ja01557a022 - 芳香取代反应。XVI.1傅克反应Isopropylation苯和甲苯异丙基溴化和丙烯
乔治•a . Olah西尔维娅h .洪水,斯蒂芬·j·库恩Maryanne e·莫法和尼娜Overchuck
美国化学学会杂志》上1964年,86年(6),1046 - 1054
DOI:1021 / ja01060a016 - 芳香取代反应。XVIII.1傅克反应t-Butylation与叔丁基溴苯和甲苯和异丁烯
乔治•a . Olah西尔维娅h .洪水,和玛丽·e·莫法
美国化学学会杂志》上1964年,86年(6),1060 - 1064
DOI:1021 / ja01060a018 - 芳香取代反应。XIX.1傅克反应Isopropylation t-Butylation Halobenzenes
乔治•a . Olah西尔维娅h .洪水,和玛丽·e·莫法
美国化学学会杂志》上1964年,86年(6),1065 - 1066
DOI:1021 / ja01060a019 - 芳香取代反应。Isopropylation XXV.1傅克反应选择性的苄基化作用,t丁化作用的苯和甲苯
乔治·a·Olah和尼娜Overchuk
美国化学学会杂志》上1965年,87年(24),5786 - 5788
DOI:1021 / ja00952a047
本文第一句话的注意:“傅克烷基化是臭名昭著的不可靠的动力学行为”。这是因为他们在很大程度上是异构的,或发生在两个阶段。同时,脚注说明调查,应向西储大学化学系,克利夫兰,哦,之前合并,成为凯斯西储大学。 - 芳香取代反应。第二十八章。亲电芳香替换机制
乔治·a·Olah
Acc。化学。Res。1971年,4(7),240 - 248
DOI:10.1021 / ar50043a002
一个帐户Olah教授他开展工作研究各种类型的机制的亲电芳香取代反应,硝化卤化傅克酰化和烷基化作用。 - 芳香取代反应。37章。氯化锡和铝催化萘与傅克烷基化烷基卤化物。分化的活动和热力学控制产品成分和烷基萘的异构化
乔治•a . Olah和朱迪思Olah
美国化学学会杂志》上1976年,98年(7),1839 - 1842
DOI:1021 / ja00423a032
这是一个类似论文Olah教授和他的妻子Judith Olah傅克烷基化的机制,除了使用萘代替苯。萘是不同,因为有两个网站的单基取代- a和b的位置。 - 傅克烷基化与甲苯苯甲醚及其比较。主要昊图公司- - - - - -帕拉替换下的动力学条件和热力学异构化的影响
乔治•a . Olah朱迪斯·a . Olah, Toshiyuki高手
美国化学学会杂志》上1984年,106年(18),5284 - 5290
DOI:1021 / ja00330a042
摘要一个令人惊讶的一句话:“没有苯甲醚的烷基化的系统研究,然而,然而报道。因此我们进行了这样一个研究报告的结果”。有时科学这些唾手可得,彻底阅读文献可以引导你。以下论文与脱烷基化作用有关,异构化,或转让傅克反应条件下烷基化:傅克酰化: - DESOXYBENZOIN
f·h·艾伦和w·e·巴克
Org。Synth。1932年,12、16
DOI:10.15227 / orgsyn.012.0016
一个相当代表傅克酰化的实验过程有机合成著名来源可靠、独立测试实验室有机合成实验过程。产品desoxybenzoin今天称为dihydrochalcone上会更好。 - 芳香取代反应。第二十二。乙酰化作用的苯、烷基苯,Halobenzenes Methyloxocarbonium (Acetylium)具有Hexachloroantimonate
乔治·a·Olah斯蒂芬·j·库恩西尔维娅h .洪水和芭芭拉难的
美国化学学会杂志》上1964年,86年(11),2203 - 2209
DOI:1021 / ja01065a020
摘要芳烃与预制CH的乙酰化作用3有限公司+盐,Olah教授如何使用SbF隔离5。同时,注意这些论文提交从——Olah教授从匈牙利在1950年逃到加拿大和加入陶氏化学公司。 - 芳香取代反应。制造。傅克酰化取代的苯和甲苯酰卤化物。取代基位置和选择性的影响
乔治•a . Olah和Shiro小林
美国化学学会杂志》上1971年,93年(25),6964 - 6967
DOI:1021 / ja00754a045
这是一个机械的研究傅克酰化(或“苯甲酰化”在这种情况下)使用哈米特的方法,一个典型的工具在物理有机化学。更多的亲电试剂反应较低kt/ kb率和低o / p选择性,而亲电试剂反应有更高的更少kt/ kb比率和高o / p选择性。kt/ kb在这种情况下是指的相对速度与甲苯和苯的反应。氧化(或oxyfunctionalization)烃和芳烃是绝对有可能正确的条件,如以下两篇论文描述。乔治Olah教授写了一系列的论文。bdapp平台 - Oxyfunctionalization的碳氢化合物。8。亲电的苯羟基化、烷基苯和halobenzenes超强酸的过氧化氢
乔治•a . Olah和Ryuichiro产生
《有机化学》杂志上1978年,43(5),865 - 867
DOI:1021 / jo00399a014 - Oxyfunctionalization的碳氢化合物。14。芳烃亲电羟基化与双(三甲基硅烷基)过氧化氢/ triflic酸
乔治·a·Olah和托马斯·d·恩斯特
《有机化学》杂志上1989年,54(5),1204 - 1206
DOI:1021 / jo00266a041
摘要双(三甲基硅烷基)过氧化物(TMSOOTMS)用作氧化剂。
谢谢你回答我的问题!
我有一些怀疑的机制引起的傅克反应的两倍。机制,三氯化铝攻击氧原子的羰基氧原子取代环的一部分,这是不同于无水三氯化铝不进攻羰基氧原子如何当我们使用酰卤化物。机制有一个原因是不同的在这种情况下吗?而且,如果我们使用相同的环氧环,但没有羰基氧原子,傅克反应还会发生吗?
另外,三氯化铝催化剂,我们应该回到三氯化铝的反应。然而,在你的机制,我们[OAlCl3]−代替。所以,不该碳链上的氧原子依然存在,导致形成羧酸之前发生碳链的环化?
这就跟你问声好!我有两个问题:
1)指出,碳正离子重排可能发生傅克烷基化反应。然而,这似乎只发生在三氯化铝作为催化剂。当FeCl3用于给定的例子,茴香素变成一个小产品,丙基苯变成主要的产品。为什么发生这种情况吗?
相关链接:https://tinyurl.com/4jy6kfea,https://tinyurl.com/yjm9smc9
在这个问题(2)https://tinyurl.com/bddnkd97)的反应P, Q,我不确定如何三氯化铝高度环氧和由此产生的傅克反应发生,我想知道你是否可以解释这种机制如何发生?
1)我不认为有什么神奇之处FeCl3。弗里德工艺加热就会发生重组。
2)你好,是的,我画出了这双傅克酰化反应的机制。https://cdn.masterorganicchemistry.com/wp-content/uploads/2022/08/Supp-1-Mechanism-for-intramolecular-Friedel-Crafts.gif
有机化学2:第一学期的课程概念回到咬你屁股。™詹姆斯先生请不要把这样的句子。
我偶尔可以无礼。
谢谢你的关心我的查询…请告诉我的角色是什么Anhy三氯化铝在FC苯甲酸的反应吗?做三氯化铝结合羧基…如果以何种方式…很高兴接到你的电话…
请告诉我明确的机制,为什么苯甲酸不接受弗里德尔工艺品烷基化和酰化反应…谢谢你…。
弗里德工艺反应一般失败当电子撤回组存在环。羧酸是一个电子撤回,这使芳环少电子丰富(因此亲核性较弱)。
我学习非常满意你的有机化学概念…真正的掌握有机化学有机化学硕士我话题方便…半岛赛事体育
我请求你,作为一个老师要我用你的内容对我的内联教室教学…期待你的建议…祝你有美好的一天
我是jee野心家和你的文章内容和精美解释……。谢谢你
你在酰化和烷基化机制,三氯化铝的产品列表,这意味着催化反应。如果是这样,为什么文学俱乐部的反应总是使用化学计量或过多氯化铝吗?
报价:“把这个“可能不需要知道类别”,但傅克酰化催化剂营业额不是很好。在“现实生活”,一个化学计量所需的三氯化铝通常是由于强烈三氯化铝坐标酮产品。”
我不包括无水三氯化铝的酮协调计划,因为我想让他们尽可能简单,整洁。
产品如果C6H5 + FCH2Cl (BCl3路易斯酸)- > ?
什么是更好的离去基团,Cl或F ?
请包括使用其他类型的苯分子的例子,尤其是代替。
谢谢你的建议。对于这篇文章我想要只使用简单的例子。我建议去一些其他的文章合成一些稍微复杂的例子。反应指南也有更复杂的例子。
嗨。你怎么知道哪些组将禁用一个苯环,傅克反应不会工作?这是所有去活化组除了卤素吗?我努力寻找一个包罗万象的规则。这些群体不同烷基化和酰化的fc ?什么组织以外的氨基,NHR, NR2强大到足以防止傅克反应吗?
有两个因素。首先,团体比卤化物将使环电子撤回停用,东亚峰会不会发生。这个刚刚从巨大的试错,说实话。第二,如果组织存在,可以作为亲核试剂(胺是最突出的)他们将与傅克反应反应而不是芳环!出于这个原因,如果你的芳环包含一个胺,保护它作为一个酰胺和删除它。
我想知道弗里德工艺适合CF3COCl酰化。
芳环与电子捐赠集团被激活。
两个注意事项:
小:错过一个负电荷AlCl4在步骤3中酰化当它作为一个基地。
主要:当你讨论减少方法酰化的产物,你列表Kishner-Wolff Clemmensen, *和*在钯氢吗? ? ?方法删除C = O。最后一个只会减少酒精。如果你想使用H2 / Ni(不是仍然Pd),你需要把它变成thioacetal第一。
谢谢你的纠正打字错误!苄基的酮加氢工作(并将减少苄醇),但我不会做脂肪族酮类。
相比之下,兰尼镍还原的thioketal好心提到应该几乎任何类型的酮。
像往常一样,好的文章。不过,需要一些小的修正:
——“最终的结果是,协调路易斯酸的亲电试剂的物种是一个更好的亲电试剂。“应该读“…使物种一个更好的亲电试剂。”
——“这是第一步进行傅克酰化反应苯和1-propylchloride之间…”应该读:“…之间的反应苯和丙酰氯,…”
——再加上一些其他小的东西,比如双或不必要的空间,等等。
非常感谢。固定的拼写错误。我在你的债务!