置换反应
SN1机制
最后更新:2023年2月7日|
的SN1反应机理
- 有两个亲核取代机制——的重要课程SN1和SN2机制。
- 的SN1的机制是不同的SN2以几种不同的方式。
- 反应是最快的三级烷基卤化物和慢主要和甲基卤化物
- 法律是率单分子,只有依赖的浓度底物(即。烷基卤),而不是亲核试剂
- 烷基卤化物与手性中心”α碳“将产品提供的混合物保留的配置和反演的配置。(注2有时这被描述为“外消旋化”。
- 这个反应是如何工作的最好的解释是,它开始于一个(速度决定)的损失离去基团给一个碳正离子弱,从而进行攻击亲核试剂在脸上,导致立体化学的损失。
- 的SN1有时伴随着碳正离子重排反应。
表的内容
1。SN1反应的立体化学:保留和反演是观察到的混合物
如果我们从一个光学纯的产品,(也就是说,一个对映体),这些反应会导致产品的立体化学的混合物是一样的起始物料(保留)或相对(反转)。换句话说,某种程度的外消旋化将(看到帖子:外消旋混合物是什么?)
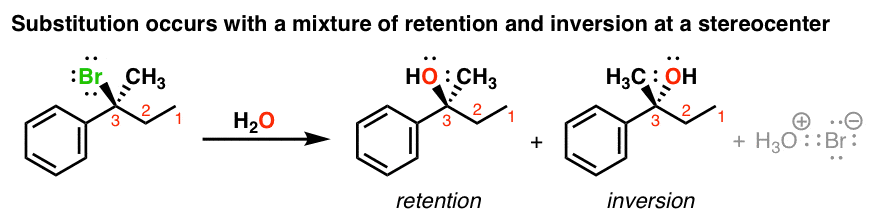
比较这个年代N2,它总是在反演结果的立体化学!显然不同的东西必须。
2。SN1反应的速率定律是一阶
我们也可以衡量这些反应的速率定律。当我们这样做时,我们注意到率只有依赖于底物的浓度,但是不的浓度亲核试剂。
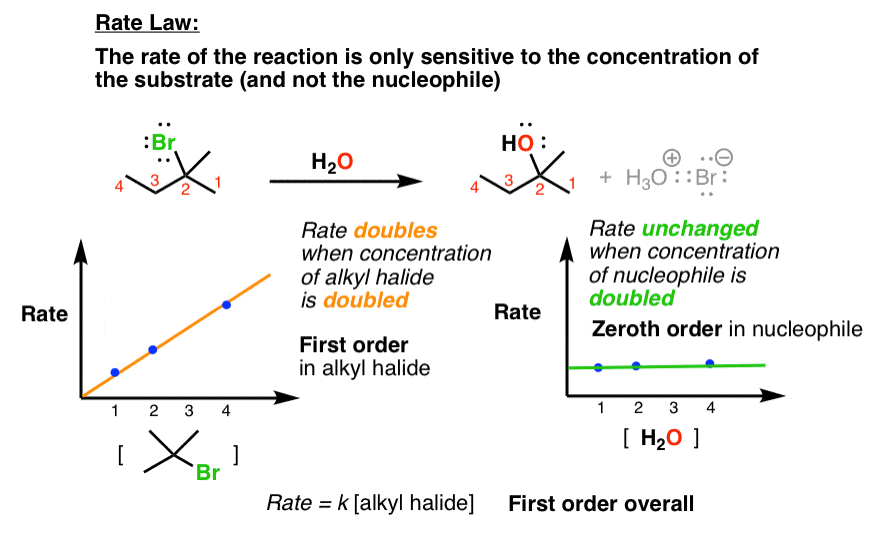
奇怪的。请记住,年代N2取决于两种。为什么这个反应只有取决于基质的浓度?
3所示。在碳反应速率增加而替换
当我们巧妙地改变基质的类型(如。烷基卤化物)我们使用在这些反应中,我们发现三级基质(例如,t溴化丁基)快比二次烷基陈词滥调,反过来速度比主(注1]
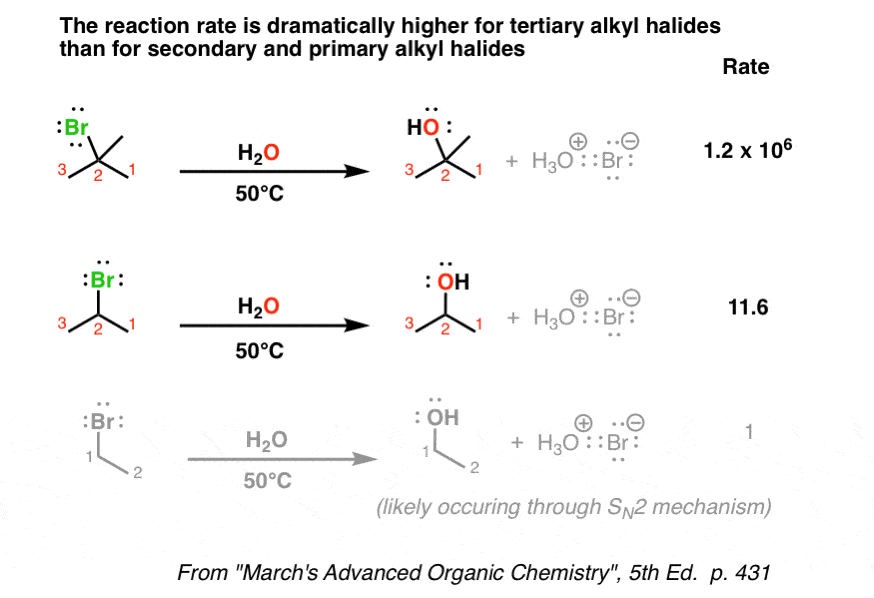
相比之下,年代N2,主要是速度比二、三级几乎没有反应。神秘的!
4所示。的逐步反应机理N1反应
最好的假设我们有这个反应是一个分段机制。
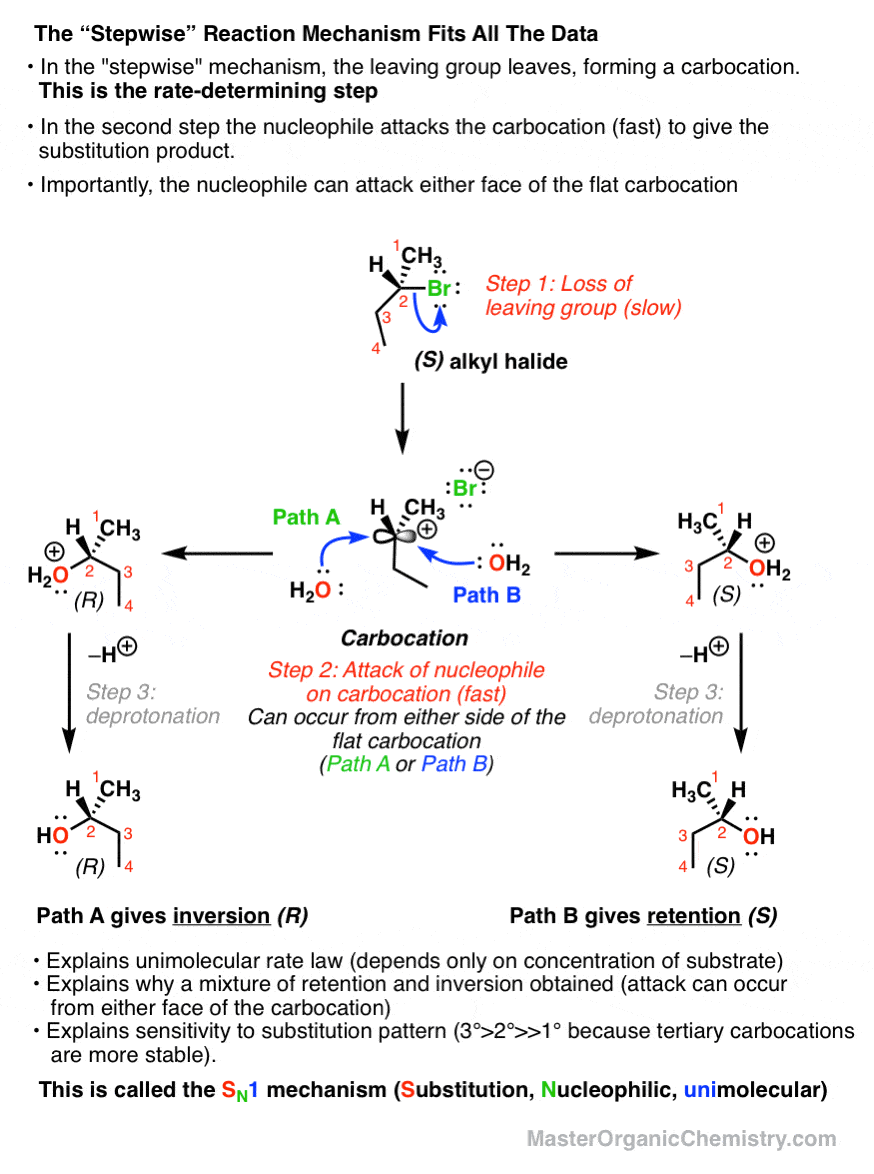
这就解释了我们所有的观察。首先,慢一步应该(不稳定)的形成碳正离子只——这取决于衬底,不是亲核试剂。
此外,自碳正离子的稳定性很大程度上取决于替代模式(三级比二级碳正离子更稳定,比小学更稳定)这也顺便解释了反应速率的依赖在替换模式(看到帖子:碳正离子的稳定性)
稳定的碳正离子,任何因素增加的速率离去基团可以离开。
它还帮助我们理解立体化学。自亲电试剂是平的,攻击可能发生从脸;这意味着我们得到保留和反演产品的混合物。
这是因此被称为年代N1机制——亲核取代单分子与SN2(替换,亲核、双分子的)。
这一切似乎如果你有一个好的工作离去基团(如卤素)。但是,如果你没有一个好离去基团吗?在下一篇文章我们将讨论如何使一个贫穷的离去基团一个好的。
笔记
注1。——主烷基卤化物这里肯定是反应仅仅通过一个年代N2机制。
注2。虽然人们常说的年代N1收益与立体的“外消旋化”,在实践中50/50分割获得的立体可能不是由于“离子配对”效应。
换句话说,离去基团可以离开,但从碳正离子的附近没有完全分离,这可能阻止亲核试剂从攻击亲电试剂的脸。因为这个原因有点更正确的说它收益“保留和反演的混合物”,而不是“外消旋化”。
(高级)引用和进一步阅读
-
56。饱和碳原子的替代机制。诉部分水解的叔。氯化丁基。
爱德华·d·休斯。
j .化学。Soc。1935年,235年
DOI:10.1039 / JR9350000255
原始研究的水解t氯化丁基被发现一阶烷基卤化物和零阶的基础,我们现在知道的机制SN1。 - 饱和碳原子的替代机制。第九部分。的一阶水解溶剂的作用烷基卤化物
莱斯利·c·贝特曼和爱德华·d·休斯
j .化学。Soc。1937年,1187 - 1192
DOI:10.1039 / JR9370001187
水解的烷基陈词滥调在甲酸水,相对利率100°MeBr 1.00, EtBr 1.71, 44.7 iPrBr, tBuBr ca。10 ^ 8。 - 反应动力学和瓦尔登湖反演。第一部分均匀水解和醇解β-n-octyl卤化物
爱德华·d·休斯,克里斯托弗·k·英格尔德和Standish家长
j .化学。Soc。1937年,1196 - 1201
DOI:10.1039 / JR9370001196 - 反应动力学和瓦尔登湖反演。第四部分。行动hydroxylic银盐的溶剂β-n-octyl溴和氯α-phenylethyl
爱德华·d·休斯,克里斯托弗·k·英格尔德和Standish家长
j .化学。Soc。1937年,1236 - 1243
DOI:10.1039 / JR9370001236
这两个文件检查仲辛卤化物的反应为了看纯粹的年代N1或年代N2通道在同一衬底可以支持仅仅通过改变反应条件。 - 溶剂分解率的相关性
欧内斯特·格和美国Winstein
美国化学学会杂志》上1948年,70年(2),846 - 854
DOI:1021 / ja01182a117
这是一个非常重要的论文,首次讨论“Grunwald-Winstein方程”。这个方程是哈米特的扩展方程,以溶剂效应(即电离能力)考虑在内。 - 金刚烷的桥头堡化合物的反应
保罗·冯·r·Schleyer和罗伯特·d·尼古拉斯
美国化学学会杂志》上1961年,83年(12),2700 - 2707
DOI:1021 / ja01473a024
桥头堡碳正离子通常是很不稳定的,因为他们不能达到所需的平面几何hyperconjugative稳定好。有点令人惊讶的是,在本文中发现SN1慢反应1-bromoadamantane收益只有1000倍比叔丁基溴化,尽管只有保留配置(当然)。 - 分子内重排的共同基础。VI.1新戊二碘化反应
弗兰克·c·惠特莫尔·e·l . Wittle和a . h . Popkin
美国化学学会杂志》上1939年,61年(6),1586 - 1590
DOI:1021 / ja01875a073
一纸证明年代N1反应可以通过反应的诱导烷基卤化银盐。在这种情况下,新戊基阳离子迅速将更稳定t戊基阳离子,这些产品。 - 饱和碳原子的替代机制。第29一部分。位阻的作用。(D)部分的机制与水乙新戊基溴的反应酒精
即Dostrovsky和e·d·休斯
j .化学。Soc。1946年,166 - 169
DOI:10.1039 / JR9460000166
这是一个例子的年代N1与重排反应。新戊基溴化水乙酒精给了t戊基酒精(和t戊基乙基醚)。 - 饱和碳原子的替代机制。第45一部分。温度对单分子之间的竞争的影响溶剂离解作用和氯化di-p-tolylmethyl non-solvolytic替换。激活快一步的单分子non-solvolytic替换
奥黛丽r . Hawdon e·d·休斯和c·k·英格尔德
j .化学。Soc。1952年,2499 - 2503
DOI:10.1039 / JR9520002499
它可以运行N1反应的补充道亲核试剂,如水解的苄氯化物的叠氮化钠。单独的叠氮化碳正离子和生产的形成可以被测量。 - 氢甲醇分解的光学活性的2,4-Dimethylhexyl-4-phthalate
冯·e·林根和哈罗德·h·蔡司
美国化学学会杂志》上1953年,75年(19),4733 - 4738
DOI:10.1021 / ja01115a035
一个早期的例子N1反应没有完整的外消旋化。林根教授提出一种机制,有趣的阅读。 - 第四纪stereocentres通过enantioconvergent催化N1反应
Wendlandt, A.E.、Vangal p &雅各布森,“
自然556年447 - 451 (2018年)
DOI:1038 / s41586 - 018 - 0042 - 1
这是一个罕见的例子,一个不对称的年代N通常1反应- SN1反应是教会给非手性的产品,但在这种特殊情况下可以诱导手性,因为碳正离子高度稳定(三级、苄基的和丙炔)。
如果亲核攻击后,碳不是一个手性中心,然后不会只是给我们保留配置产品?
对叔丁基溴化去叔丁基醇,没有形成立体异构体的可能性,所以保留或倒置的问题成为一个问题。
我仍然学习亲电子/琳反应,和你的网站是一个祝福!我有一个问题关于快速方法来确定SN1或SN2反应。假设Br -离去基团。据说如果一个亲核取代反应酸(在这种情况下,《哈佛商业评论》)作为其产品,然后SN1反应。另一方面,如果一个亲核取代反应阴离子(在这种情况下Br -),那么它的SN2反应。我确定这个方法是有用的和启发式,但无论是SN1或SN2,不该离去基团Br -不能形成哈佛商业评论(这是一个软弱的共轭碱)?我认为你写H3O + Br -显示这个,但我可能是错的。请给一些澄清吗?谢谢你!
你可能只看到哈佛商业评论如果你执行替换醇反应。目的是把可怜的离去基团哦好的离去基团水,然后,根据底物是否主要(SN2)三级(SN1)或二级(问问你老师,回答不同!)你将获得由此产生的替代产品。
嘿,詹姆斯,我最近遇到本文:http://dx.doi.org/10.1021/ed086p519
这是否意味着SN1机制并不适用于二次烷基卤化物吗?如果是这样,请发表一篇文章解释为什么(我还是一个学生,和并不真正理解纸)
这是一个非常有趣的纸。
我不会说SN1并不适用于二次烷基卤化物* *让我们只是说他们比他们似乎不太重要。
假设整个教学SN1 SN2 / E1和E2非常混乱,这样已经持续很长一段时间。如果你把介绍有机你会认为有机化学家们花了大量的时间在计划SN1 Sn2 / e1和e2反应而在现实的比率发表Sn2 vs SN1和e1和e2反应反应至少100:1。即使这样SN2最好在主基板,以避免副反应。
这是一个烂摊子。我很高兴有人回去和透过原始英格尔德/休斯报纸,因为他们很多是什么教的基础。
动力学可以提供所有必要的细节亲核取代反应?
我不确定细节你所说的“所有必要的细节”,但即使是有限的阅读,忽视立体化学等问题,等等。所以答案是否定的。
可怕的和有用的内容——具体的错误是在# 3。类型的基质* * *
谢谢!
没有看到它。有人知道吗?
谢谢你的非常有用的网站!
我有一个问题关于SN1反应的第一步——离去基团的叶子。导致离去基想要离开的原因是什么?是这个动作,在热、光或其他能源来源?
谢谢你的帮助。
最好把债券作为球和弹簧。在任何给定的时刻之间的债券离去基团的碳和振荡两球由弹簧连接和键长之间的交流是比正常的要短,超过正常。一般是“键长”。
现在想象的能量债券延伸到其最长长度。碳将带部分正电荷(δ+)和离开集团将带部分负电荷(δ-)。如果碳能够稳定正电荷很好(如一个叔碳正离子)和离去基团能够稳定负电荷很好(如弱碱)然后不难想象债券长度越来越长,直到超越我们考虑普通债券的长度,可以认为是碳正离子附近的离去基团。如果一个亲核的溶剂(如CH3OH)左右它可能会“陷阱”,自由碳正离子,导致取代反应。
相反如果我们有这样的情况中,我们有一个糟糕的离去基团(OH)连接到一个碳不能够稳定正电荷(主要),那么该债券的能量屏障长度得到足够长的时间导致离解太高了。
在sn1 rexn最通常外消旋混合物形成的产品,但不是100%的外消旋,为什么?
原因是生成的碳正离子与离去基团能形成所谓“离子对紧张”。在紧张的离子对离去基团没有完全分离,这将阻止碳正离子的脸与离去基团。
为了完整的外消旋化发生需要有完整的离解,要么面对碳正离子都有同等的被攻击的概率。
请你写一本教科书——我愿意买它!
现在还没有计划,但谢谢你的信任投票!
我不能告诉你如何出色地每一个主题是解释说。这是伟大的。你的类比和措辞我理解有机化学的唯一途径。由于一吨
很高兴听到它Anisha !
我得到了它。谢谢你!
在段落在“最后注意”,我相信“换句话说,离去基团可以离开,但不是完全游离碳正离子的附近,这可能阻止攻击亲核试剂的亲核试剂,脸”应该读“阻止攻击亲电试剂的亲核试剂”。同时,在段落在“这里可能是怎么回事?”, I believe “Since the nucleophile is flat, attack could occur from either face; which means that we obtain a mixture of retention and inversion products” should read “Since the electrophile is flat” instead.
先生请告诉我倒的百分比和retented prodct在离子对机制…? ? ?
这将取决于多种因素,如温度、溶剂、离去基团和衬底的身份。高度可变的!
极大的帮助!内容非常简单,可以理解!感谢!这个网站将用于整个学期! !
是的…非常有用但有机化学非常信任化学化学< 3的其他分支
您已经创建了一个很棒的网站。我还没有使用你的反应指导太多,但我仍然像一个成员仅仅是因为我感觉你完全配得上我的钱的所有帮助我的博客。(教我比我的书都在几天内!)
无论如何,只是想指出,在你主演注意底部,我想你意味着主要卤代烷反应当然是通过SN2机制(不是SN1)。
是的,你是对的。谢谢你的捕捉,很高兴你觉得它有用!
非常有用的网站,非常感激你做出的努力,请正确的拼写碳正离子在第四行机制。
谢谢你!