二烯烃和MO理论
π成键和反键轨道
最后更新:2023年1月8日|
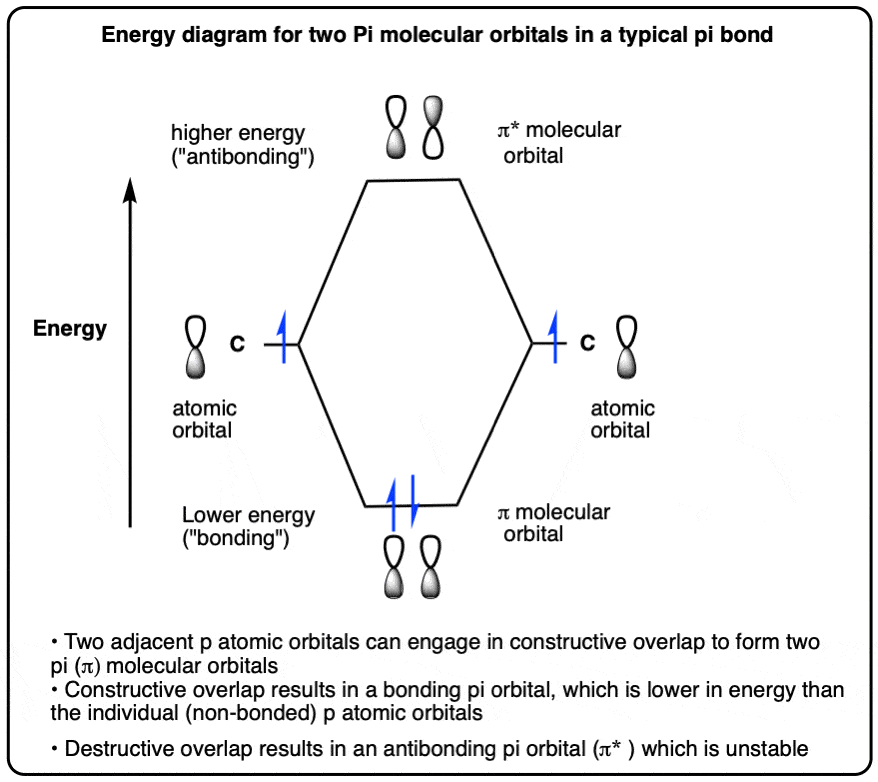
成键和反键轨道为一个简单的π键
- 两个相邻的p轨道包含一个电子可以重叠形式π键
- 在一个π键,这两个概念原子轨道(p轨道)重叠,形成两个分子轨道(π轨道)
- 两个p轨道之间重叠导致相同的阶段有建设性的轨道重叠,并允许电子两个原子之间的共享,导致成键和一个降低的能量
- 两个p轨道之间的重叠相反阶段的结果具有破坏性的轨道重叠和防止电子被两个原子之间的共享。两个带正电荷的原子核之间的斥力被紧密的结果在一个更积极不稳定情况被称为反键(π*)
- 对于一个简单的和两个电子π键,键π轨道(π)高分子轨道(人类)和反键轨道最低未占据分子轨道(LUMO)
表的内容
- 能源关系图:“结合”和“反键”
- 完整的能量图的关系
- 孤对电子,成键和反键轨道
- 能量图包括焊接、非成键和反键轨道
- “为什么反键轨道存在”?
- 分子轨道是由原子轨道的重叠
- 结合π分子轨道形式通过建设性的p轨道的重叠的旁边
- 反键π分子轨道由相邻的p轨道的破坏性重叠的旁边
- 的能量图简单的π键结合成键和反键π分子轨道
- 结论:一些规则π分子轨道
- 笔记
1。能源关系图:“结合”和“反键”
在本系列以前的文章中,“在结合和共振”,我警告说,为了获得更深入的理解的一些概念,我们需要进入MO理论。
但是在我们做之前…我们谈论约会吗?情人节是我写这篇文章时,毕竟…
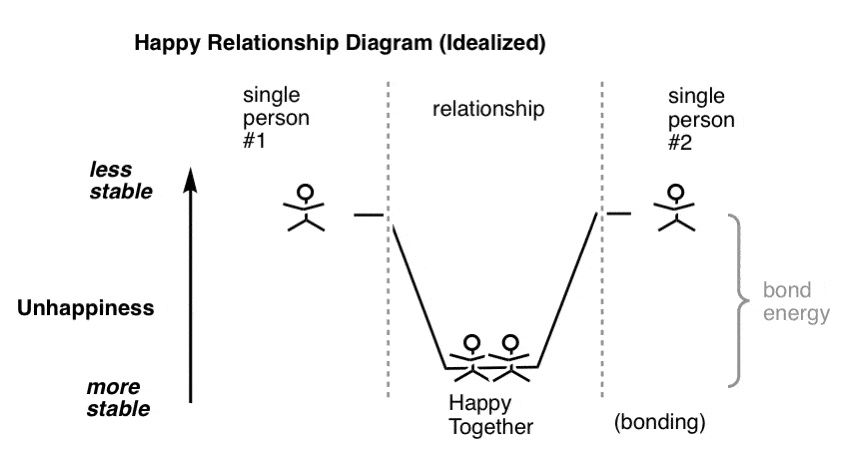
很多人说他们快乐的单身,我相信许多可能。但在他们的脑海中许多单身人士认为如果他们只是找到合适的人,他们甚至可能——或者更幸福少不开心,这是一个蹩脚的方式来看待心理但必要的如果你想画一个图,一个“快乐的夫妇”占据了“势能”。
(也可以yenta-catalyzed)
减少所带来的不快键可以量化为“键能”。
当然,它可以把大量的随机碰撞前两个单身的人落入势能,分类一个快乐的关系。
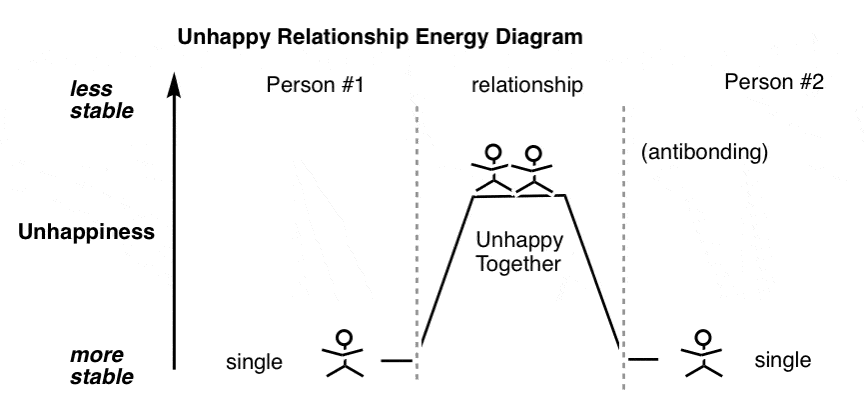
更糟糕的是,两人会突然发现自己在一段关系最初是彼此吸引,但在近距离时意识到他们实际上是快乐的单身。
给你一个像下面这样的情况,你能想到的“键”——一个不高兴的,不稳定的夫妇,注定要回到它的单个组件分离。
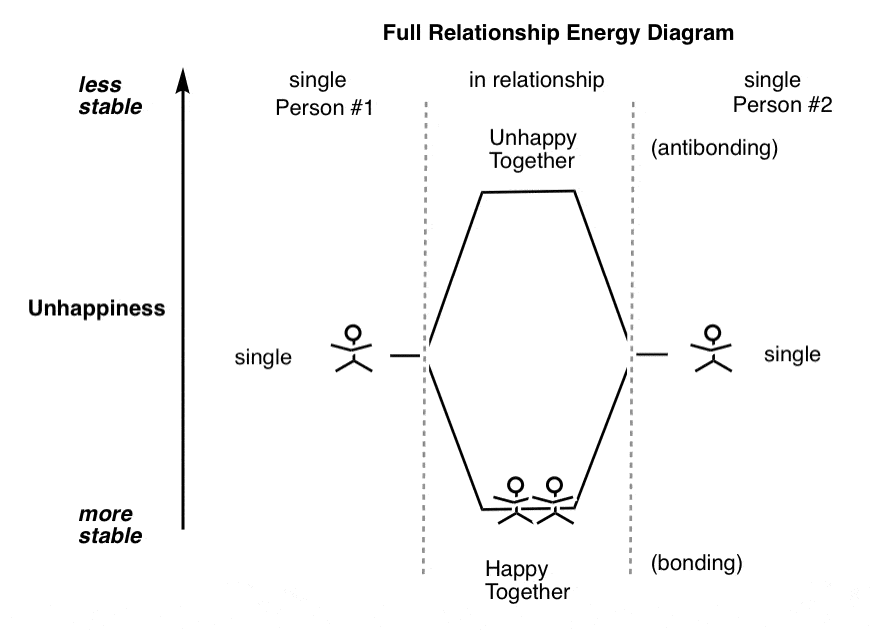
2。完整的能量图的关系
浪漫的爱的观点是,如果势能足够深,你有幸福快乐的生活,情人注定永不分离。
当然,这在一定程度上反映了这样一个事实,浪漫喜剧在婚礼现场。他们不检查在六个月后当白马王子变成了”。从未拿着纸巾离开他的裤子放到烘干机”和白雪公主已经演变成“夫人。一直让她恶心适意的刀在厨房柜台”。
事实是,即使是拥有快乐的关系潜在的不满。应变他们有足够的力量,和不稳定(分手)可能的结果。
这图是两个人!
我们不需要做图纸的不稳定可能发生当一个第三人进入的关系。
3所示。孤对电子,成键和反键轨道
被迂腐的风险,让我们提醒自己:为什么债券形式呢?原子是什么原因会打扰彼此分享他们的电子呢?为什么他们就不能“快乐单身?”。
有些人,当然——惰性气体。但对元素的第一行元素周期表少于8个价电子,特别是碳、氮、氧、氟,有一种强烈的静电驱动力价电子的获得一个完整的八隅体。
这可能有助于回忆力在起作用。化学键,两个带负电荷的电子两个带正电的原子核之间举行。如果你使用库仑方程之间的引力模型,这两个电子和两个原子核(相反电荷吸引,切记)之间的斥力作用大于两个带正电的原子核和两个带负电荷的电子,分别。有一个网降低的能量——稳定的通过将细胞核以这种方式在一起。这种降低的能量称为键离解能。
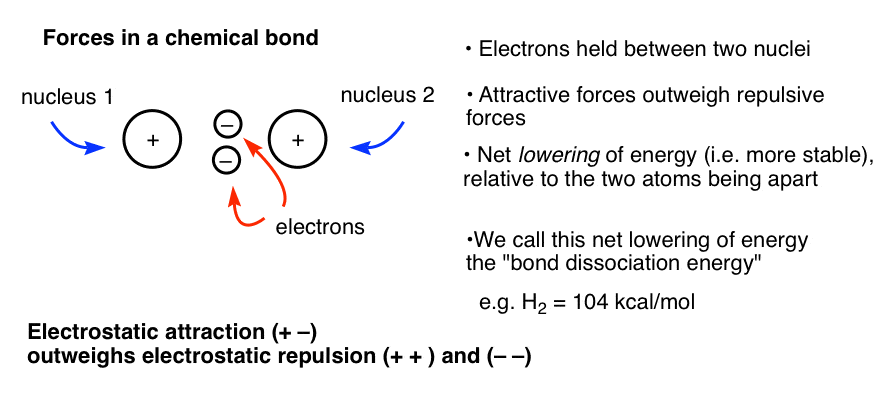
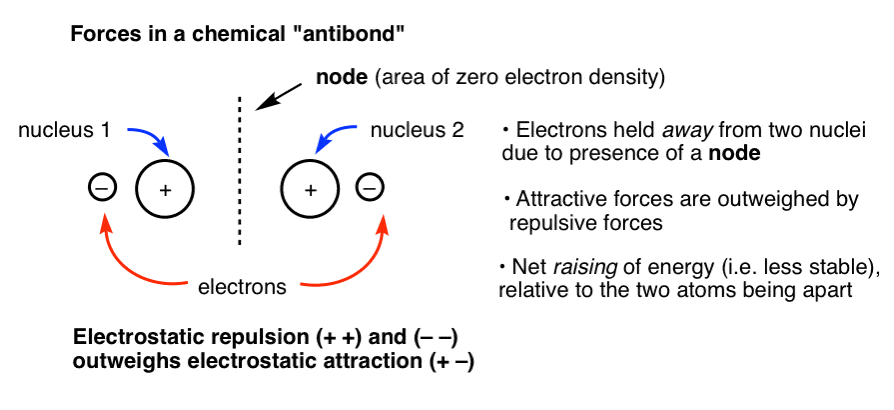
现在想象一下另一种情况,细胞核是相同的距离,但电子不能它们之间(由于举行节点,见下文)。在这种情况下,您有两个带正电的原子核在太空举行紧密但没有电子之间提供一个稳定的有吸引力的互动。两个原子核之间的斥力(和电子互相)大于任何电子和原子核之间的引力,结果比两个非键原子的情况更不稳定。我们称这种情况“键”。
4所示。能量图包括焊接、非成键和反键轨道
像的关系,在化学成键我们看到三个不同能量的情况。
- “非键”——原子的能量没有任何联系。
- “键”——快乐的能源情况之间的两个电子原子,这是更稳定:景点>排斥
- “反键”——能量不快乐的情况下举行的电子从原子:排斥>吸引力
两个相同的原子间的成键,我们可以大致勾勒出这三个层次如下。(注2]
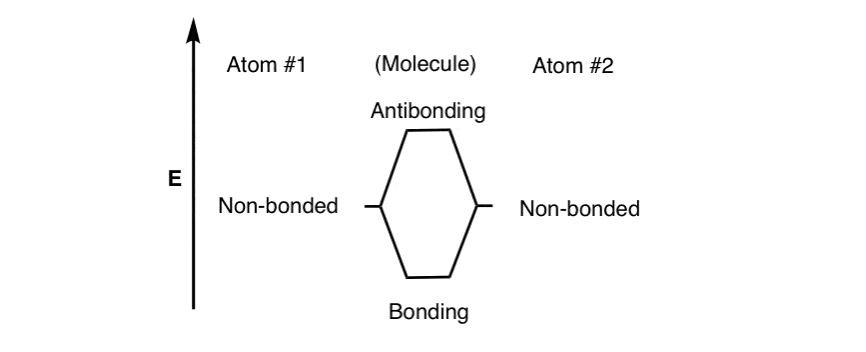
注意,我们还没有填充电子水平。下一个。
5。“为什么反键轨道存在”?
“为什么反键甚至存在吗?”,你可能会想。为什么我们要关注自己的情况比两个更不稳定的原子被分开吗?
我们会看到,它是很平常的事。电子占据反键的状态——尽管短暂时间的分子——没有被摧毁。电子小(1/1840质子的质量)和原子核是沉重的;电子可以压缩成键轨道的反键状态和回落将在更少的时间比债券分离。(注1]
事实上,每次你观察的绿叶植物或一朵花的花瓣,你看到的是可见光的结果(频率ν)被吸收发色团(如叶绿素),促进一个电子从π(粘结)轨道π*反键轨道,能量E = hν差距。高能电子可以放松回到基态,发射光子的过程中,分子回到开始的地方。
至于“为什么反键的存在”,这是一个非常好的问题。
没有电子可能完全相同的量子数。成键轨道可以容纳两个电子,每个自旋相反,就是这样。一夫多妻制可能是一件事在某些文化中,“另类生活方式”比比皆是,但沃尔夫冈·泡利不相容的不相容原理比任何宗教或文化更牢不可破的条例。
“量子数”,我们的意思是(n,l米和自旋),薛定谔波动方程的参数,描述了电子的能量和位置。我们所说的“轨道”实际上是三维形状,有95%的一个电子被发现的概率。每个轨道(一个独一无二的结合n, l,和米)可以容纳两个自旋相反的电子,+ 1/2和1/2。
因此,当我们填充与电子能量图,任何单独的轨道可以容纳的最多的是两个。
的构造原理首先指出,能量最低的轨道了。这有点像一个灰狗巴士;人们只能站在过道一次座位填满。
所以有两个电子(由half-arrows)填充轨道,我们的草图是这样的。注意相反的旋转在成键分子轨道。独占的轨道,电子自旋的方向一般是相同的(点头洪特定律)
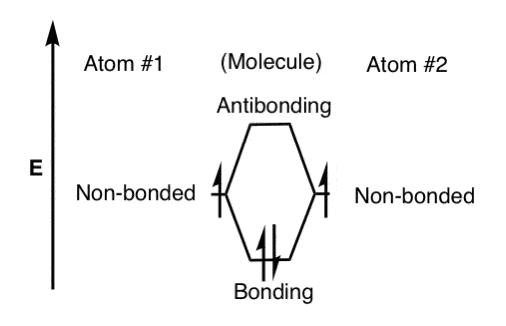
第三个电子被添加到这,我们必须把它放在一个反键轨道(不稳定)。并添加第四个电子,我们会有一个更加密集的反键轨道,这是极其不稳定的(和一个很好的描述他为什么氦不形式2)。
6。分子轨道是由原子轨道的重叠
我们通常关心的轨道介绍有机化学的年代和p轨道。
年代轨道一般有球的外观。p轨道像哑铃。d和f轨道有各种有趣的形状,我们这里不讨论。
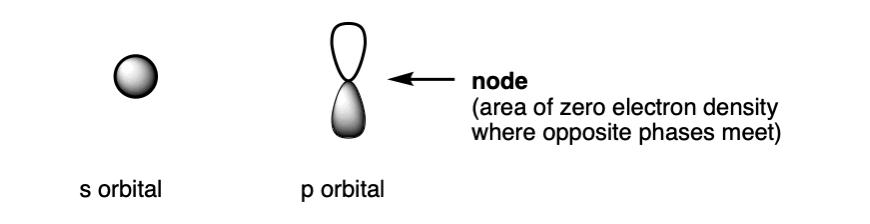
点在p轨道的中心是所谓的节点零的电子密度,之间有一个过渡阶段。2 p轨道,直接在核节点驻留。有趣的事实:节点数的增加,主量子数,n。2 s和2 p轨道都有一个节点,3 s和p 2,等等。
当原子轨道重叠时,他们的形式分子轨道:电子在原子之间共享的空间领域。
当s-0rbitals相互重叠(或杂化轨道提到,比如sp3sp2, sp),σ(σ)形成分子轨道。这是“端点的”重叠。氢的键(H2)是一个很好的例子。
从未改变轨道的成键。分子轨道的数目总是等于原子轨道的贡献。因此对H2由两个原子轨道(1),我们获得两个分子轨道(成键和反键)。结合在CH4,由4个氢原子1 s轨道和4混合碳sp3轨道,总共有8分子轨道。
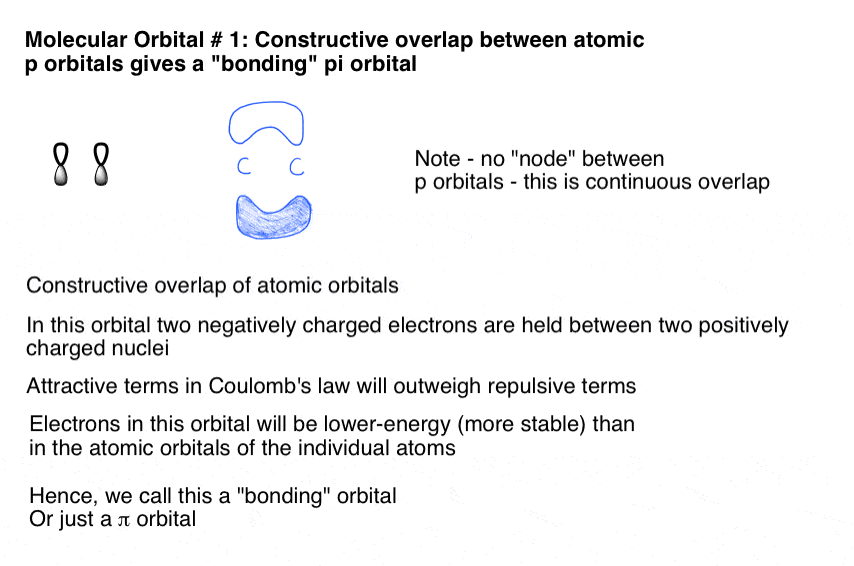
7所示。结合π分子轨道形式通过建设性的p轨道的重叠的旁边
在这个系列文章中,我们已经讨论ππ键,这是两个p轨道之间的相互作用(重叠)。键在这里不像在端点的σ键,但“侧面”,正如我们上次看到的,是负责旋转障碍在烯烃。
当两个p(原子)轨道重叠,我们形成2π分子轨道(π)。
“结合”情况描述建设性的重叠,p轨道匹配相连接在一起形成一个π分子轨道,它看起来像这样:
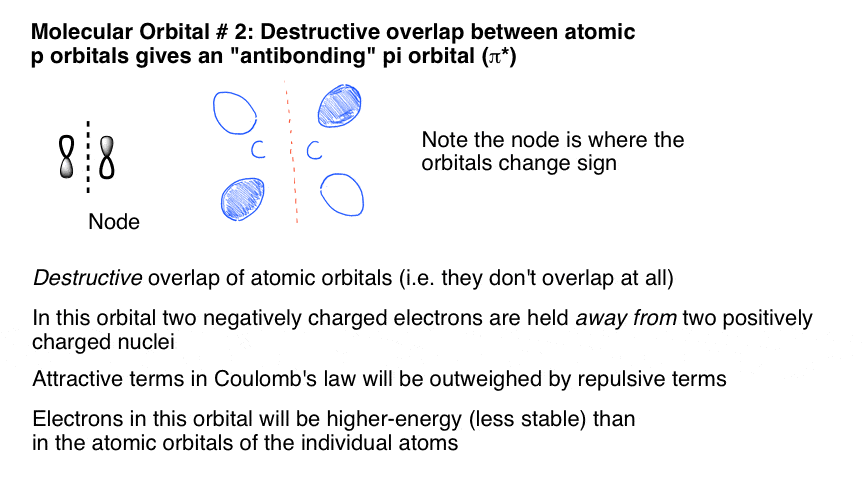
8。反键π分子轨道由相邻的p轨道的破坏性重叠的旁边
第二个分子轨道是所描述的破坏性的重叠(或者破坏性的干扰,如果你喜欢)p轨道的不匹配相连接在一起,形成另一个π分子轨道。
注意,这个结果节点原子之间——在这一领域,有一个变化阶段,因此没有轨道重叠(因此没有电子密度)。这对应于“键”情况我们前面所述,两个原子核在哪里举行紧密空间但缺乏吸引力的电子之间的相互作用。因此,更高的能量。
{kind=link}
9。的能量图简单的π键结合成键和反键π分子轨道
通过类比我们之前的例子,我们可以制定一个能量图两种情况。(反键轨道的能量是略大于2倍的结合能)。(注3]
根据构造原理,这些轨道将填补在秩序的稳定,这意味着对于一个典型的π键,我们得到两个电子ππ*轨道和零。
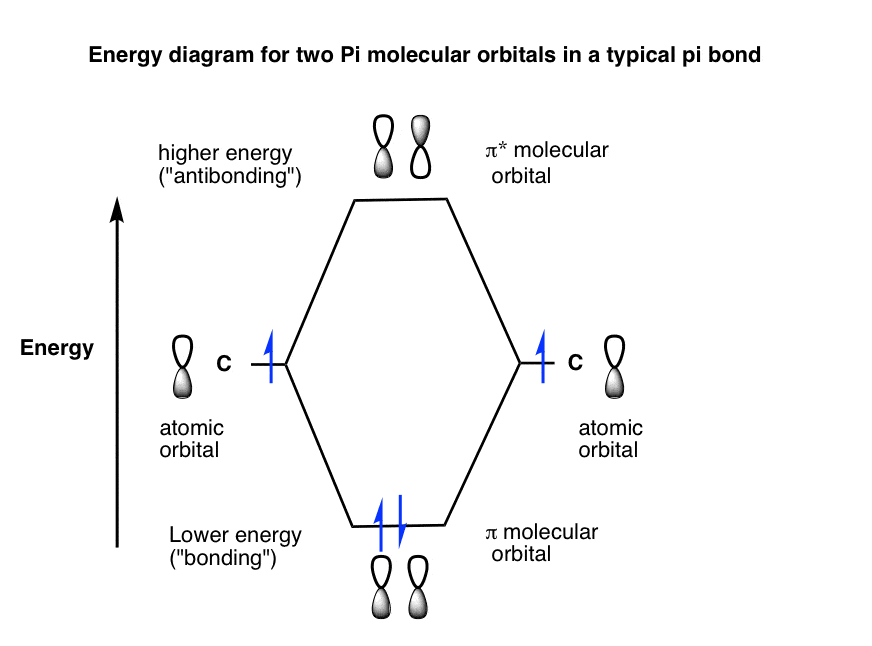
如果我们添加第三个电子,它必须去π*反键轨道。
对于简单的π键的情况下,注意,π分子轨道是“最高占据分子轨道(或HOMO)和π*轨道是“最低未占据分子轨道”,或LUMO。这不是一个重要的区别目前,但它会在后续文章。
10。结论:一些规则π分子轨道
希望这是一个大多数人从第一学期材料审查。我们将使用这个练习来形成一套准则,我们可以应用建立更复杂的系统分子轨道组成三个,四个,甚至更多的各个组件。
让我们来回顾一下要点。
- 当相邻的p轨道重叠是可能的,那么就可以形成π分子轨道(π)。(注意4]
- π分子轨道由重叠的数量总是相等数量的p轨道
(另一种方式说:重叠的N原子轨道产生N分子轨道) - 当N = 2,这个描述一个π键。
- 一个节点阶段改变符号。
- (最稳定的)最低轨道零节点之间的单独的轨道(如π成键轨道)
- 高能(不稳定)的轨道n - 1节点之间的单独的轨道。(我们的π键的例子,因为N = 2,一个节点)(
在下一篇文章我们将介绍系统的三个和四个分子轨道。
N = 3描述了烯丙基系统。我们将讨论的分子轨道烯丙基阳离子、烯丙基自由基和烯丙基离子。如果有时间,我们甚至可能得到N = 4 (butadienyl)系统。
在我们做之前,一个测试:
- 有多少分子轨道将烯丙基系统?
- 高能分子轨道会有多少个节点?
- 有多少分子轨道将butadienyl系统?
- 有多少个节点的最高能量的分子轨道?
(答案]
多亏了托马斯Struble与这篇文章帮助。
笔记
注1。这是本质的奥本海默近似出生,即原子和电子的运动可以分离。如果你想继续殴打我们的关系类比而死,甚至类似于快乐的夫妇会有争端,然后弥补刺耳的摩擦轮前的离婚律师有时间操作。
注2。两个相同的原子的能量两个原子轨道的贡献是相同的。当我们沿着周期表(增加电负性)原子轨道的能量减少,另一种说法更稳定。
注3。π*比π键,键由于电子斥力。看到弗莱明,“前沿轨道和有机化学反应”(第二章)最明显的和最好的治疗在有机化学分子轨道。
注4。对相邻的p轨道而做的一个例子不导致分子轨道,参见下面“桥头堡烯烃”。
这是两个相邻p轨道的一个例子,不能形成一个π键,如烯烃的桥头堡。系统像两个人的激进分子。
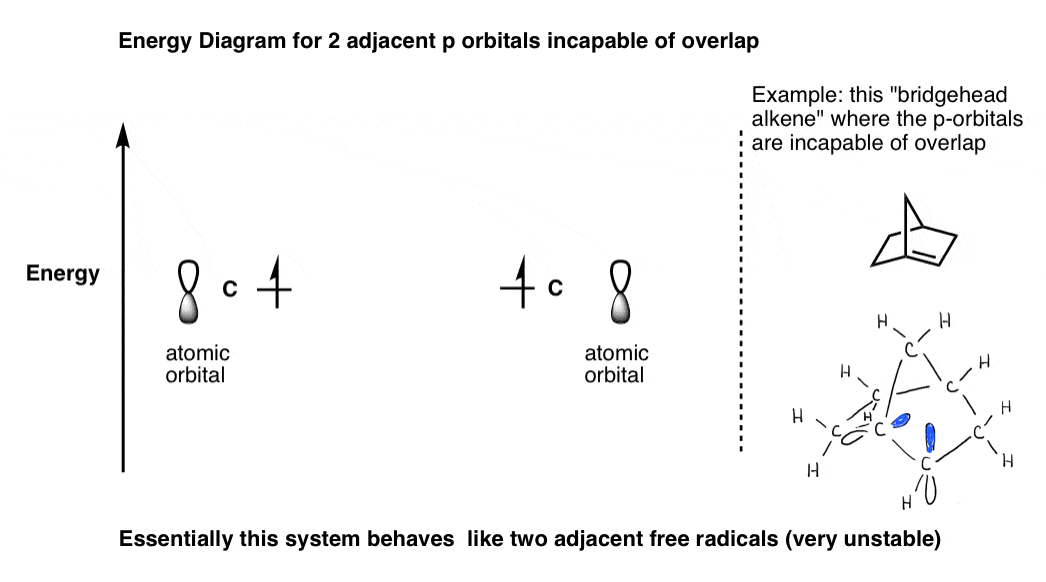
注5。烯丙基(N = 3):三个p轨道产生三个分子轨道。高能分子轨道会有2个节点(n - 1)
Butadienyl (N = 4):四个p轨道产生四个分子轨道。最高能量的分子轨道将有3个节点(n - 1)。
你好想引用的一些照片在我的博士论文我如何申请权限吗?
只是引用的URL
詹姆斯,这是一个非常优秀的文章,你——我非常喜欢单一/夫妻类比…
相比之下之上所写是头脑麻木,目的是高于你的网站的目标受众的水平——这是一个很好的例子“教育学”的类型和科学写作的那些来这里寻求避免!就我个人而言,我会删除它…
我不知道我能有这样的一个爆炸在阅读关于轨道。谢谢你的愉快的体验考试的!
很高兴它不是完全unentertaining !
苯定量(论文)。
见图(苯定量,DOI: 10.13140 / RG.2.2.33278.64328),
https://archive.org/details/BenzeneQuant,
http://vixra.org/pdf/1902.0030v2.pdf
它良好的可视化(苯定量)沸腾的正负电子对,它总是发生在化学成键的多重性。我们有一个美丽的“量子力学模式”一个苯分子与一个三电子键。我真的很喜欢:),因为它传达了意思…
“…因此,化学领域的结合,在这种情况下,一个电子不能被视为一个“point对象”,因为它(电子)将花部分时间在国家”电子+一对(正电子+电子)”,因此其交互应该描述量子场论的框架。这种方法可以解释,对于多电子化学键(twoelectron、三电子等),电子间的斥力是克服:由于化学键实际上是一个“沸腾的质量”的电子和正电子,虚拟正电子“帮助”克服电子之间的排斥力。这种方法假定化学键实际上是一个封闭的空间袋(一种潜在的能源意义上的),“沸腾”的真实电子以及虚拟正电子和电子发生,和潜在的“体积”袋实际上是一个“量”的化学键和量子力学的空间测量电子的位置的不确定性。
严格地说,这样的考虑,电子不再有一定的能量,动量,坐标,并不再是一个“质点”,但实际上占用化学键的“卷”。可以说,在化学键单个电子是没有人性的,失去了个性,事实上,它不存在,但真正有“沸腾的质量”的电子和虚拟正电子与电子的波动变化。也就是说,化学键
实际上一个单独的粒子,如前所述,semivirtual粒子。此外,这种方法可以扩展到基本粒子的结构如一个电子和正电子的:一个基本粒子在这个考虑是一个脉动真空封闭在一定空间袋,这是一个潜在的对这些波动。
尤其值得注意的是,在这个考虑,电子是强烈相互作用的粒子,因此泡利不相容原理并不适用于化学键(更多细节见部分“泡利不相容原理和化学键”),不禁止同一threeelectron债券存在的多样性为1.5”。
第103 - 101页。回顾。苯三电子债券的基础上。(泡利不相容原理,海森堡的不确定性原理和化学键)。
http://vixra.org/pdf/1710.0326v4.pdfhttps://dx.doi.org/10.2139/ssrn.3065288
苯三电子债券的基础上:
评论(138页,完整版)。苯三电子债券的基础上。(泡利不相容原理,海森堡的不确定性原理和化学键)。http://vixra.org/pdf/1710.0326v4.pdfhttps://dx.doi.org/10.2139/ssrn.3065288
目前的研究表明:
1。芳香债券是一个三电子债券在平面循环系统与一个特定的电子通过循环的相互作用。
2。目前的工作显示了不适用泡利不相容原理的化学键(页103 - 105)。
3所示。此外,一个新的理论模型提出了化学键的海森堡测不准原理(页92 - 103)。
4所示。事实上,本文研究表明,现代概念的化学键不能严格考虑理论上没错,而是定性与实证定量计算。
Bezverkhniy之上(viXra):http://vixra.org/author/bezverkhniy_volodymyr_dmytrovych
之上Bezverkhniy (SSRN):https://papers.ssrn.com/sol3/cf_dev/AbsByAuth.cfm?per_id=2828345
之上Bezverkhniy (Quora):https://www.quora.com/profile/Volodymyr-Bezverkhniy
Bezverkhniy之上(档案):https://archive.org/details/@threeelectronbond
之上,你可以在这里把这东西,但这并不意味着它是最好的地方。
MO方法和VB的量子力学的分析方法从pq的位置。
http://vixra.org/pdf/1704.0068v1.pdf
莫法和VB法使用的原则,分析了量子叠加态(pq)和方法描述量子系统几个部分组成。结果表明,分子轨道方法的主要假设(即分子轨道可以像重叠的原子轨道的线性组合)进入一个不可克服的矛盾与量子叠加态的原则。它也表明,由几个部分组成的量子系统的描述(采用量子力学)实际上在VB中禁止把方程相应的规范结构的成员方法。
利用量子力学的叠加原理,莫法和VB法进行了分析,表明他们在矛盾与量子力学。
同时,使用的量子力学描述几个部分组成的一个系统,它是显示规范结构的归因的方程是不正确的。因此,莫法和VB方法没有描述分子化学键,但实际上,很多原子(分子由描述)。量子化学计算,也就是说,在化学键是《迷失》。因此,为了“介绍”一个化学键与量子力学计算和避免冲突,建议假设MO的存在作为一种新的基本质量,描述一个特定的化学键,而不是来源于简单的结构元素。
MO方法和VB的量子力学的分析方法从pq的位置:
http://vixra.org/pdf/1704.0068v1.pdf
1。苯分子结构的基础上,三电子债券。
http://vixra.org/pdf/1606.0152v1.pdf
2。实验确认存在的三电子债券和理论依据它的存在。
http://vixra.org/pdf/1606.0151v2.pdf
3所示。一个简短的分析化学的债券。
http://vixra.org/pdf/1606.0149v2.pdf
4所示。补充三电子债券存在的理论依据。
http://vixra.org/pdf/1606.0150v2.pdf
5。three-electrone债券理论在四个使用简短的评论。
http://vixra.org/pdf/1607.0022v2.pdf
6。审查。苯三电子债券的基础上(完整版,93 p)。
http://vixra.org/pdf/1612.0018v5.pdf
7所示。量子力学方面的l·鲍林的共振理论。
http://vixra.org/pdf/1702.0333v2.pdf
8。MO方法和VB的量子力学的分析方法从pq的位置。
http://vixra.org/pdf/1704.0068v1.pdf
Bezverkhniy之上(viXra):http://vixra.org/author/bezverkhniy_volodymyr_dmytrovych
Bezverkhniy之上(Scribd):
https://www.scribd.com/user/289277020/Bezverkhniy-Volodymyr
Bezverkhniy之上(亚马逊):https://www.amazon.com/Volodymyr-Bezverkhniy/e/B01I41EHHS/ref=dp_byline_cont_ebooks_1
这个截图(与)与解释(大多数)看到这个链接。
Bezverkhniy之上(Archive.org):
https://archive.org/details/@threeelectronbond
真诚Bezverhny之上Dmitrievich。
我的ORCID iD: 0000-0002-3725-5571