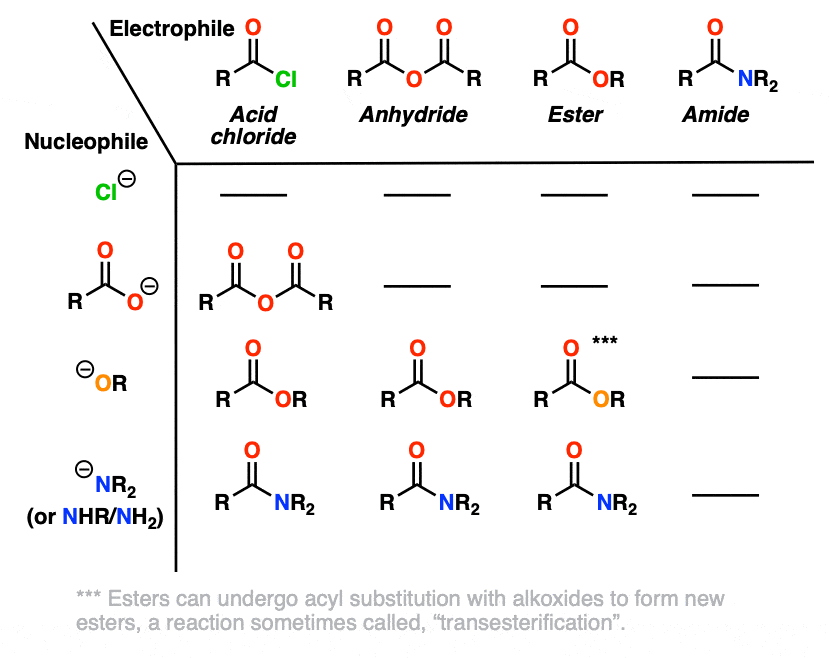
羧酸衍生物
亲核酰取代(与带负电的亲核试剂)
最后更新:2023年5月22日|
亲核酰取代(与带负电的亲核试剂)
是的,有很多的反应羧酸衍生品来学习!在本文中,我们将探索无疑是最重要的途径:亲核酰取代。
不过,好消息是:
- 与带负电的亲核试剂亲核酰取代倾向于遵循一个简单的两步机制(addition-elimination)
- 此外,对于所有意图和目的,NAS反应行为很像酸碱反应。如果你还记得,“强酸+强基地给弱酸性弱基地”,(也被称为酸碱平庸的原则)你会在你的方式能够预测是否一个给定的亲核酰取代反应将会发生。
表的内容
- 亲核酰取代(NAS)
- 三亲核酰取代反应(这工作得很好)
- Addition-Elimination机制与带负电荷的亲核试剂亲核酰取代
- 有些亲核酰替换不工作
- NAS反应时青睐离去基团比亲核试剂较弱的基地
- 一些快速练习
- 羧酸…是酸的!
- 皂化
- 分子内的亲核酰取代
- 格氏试剂和LiAlH4增加两倍
- 中性的亲核试剂和酸催化
- 总结
- 笔记
- 测试你自己!
- (高级)引用和进一步阅读
1。亲核酰取代
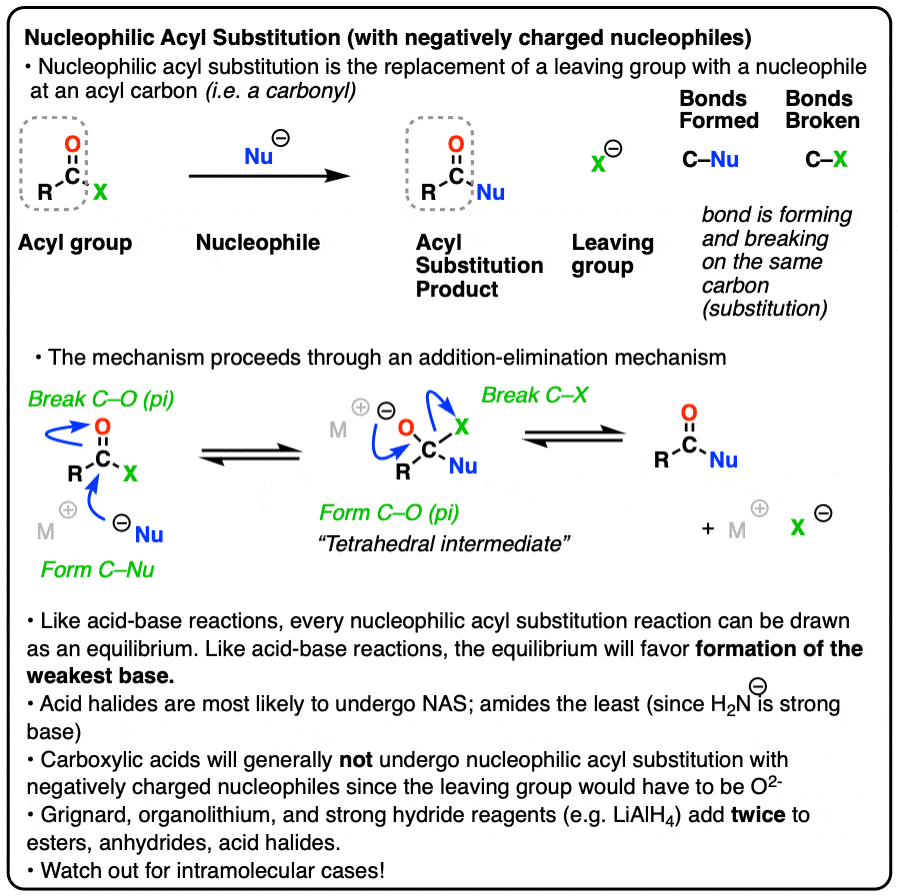
亲核酰取代是反应一个亲核试剂形成一个新的债券羰基碳酰基伴随破碎的债券之间的羰基碳和一个离去基团。
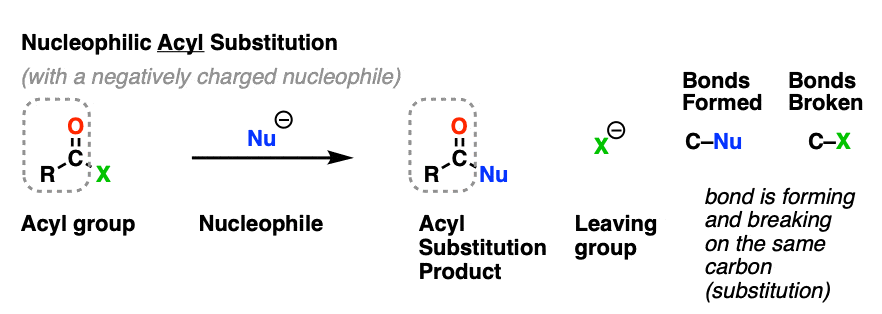
这是作为一个分类取代反应因为我们正在形成,打破债券在同一个碳。碳-亲核试剂债券形式,和碳-离去基团键断裂。
虽然机制是不同的,亲核酰取代表面上像取代反应等我们以前见过亲核的脂肪族替换和亲核性芳香取代。
{kind=link}
在本文中,我们将特别覆盖亲核酰取代反应的例子带负电的亲核试剂。在后续的一篇文章中,我们将讨论一个的情况酸催化剂采用亲核酰取代,比如费舍尔酯化,酸性水解的酯类和许多其他的例子。
(中性的亲核试剂,以及亲核取代在酸性条件下,明白了Addition-Elimination中性的亲核试剂)
2。三亲核酰取代反应(这工作得很好)
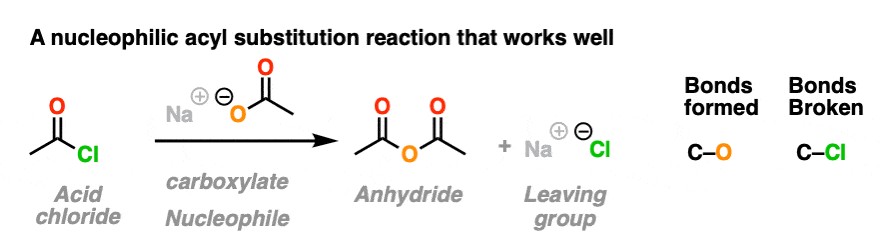
这里有三个亲核酰取代反应的经典例子很好地工作。
首先,酸性氯化物与羧化物反应(的共轭碱羧酸,充当亲核试剂在这里)给一个酐。一个新的切断键形成和C-Cl键坏了。
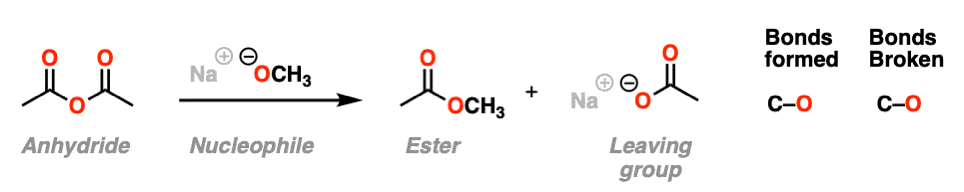
在第二个例子中,我们把一个一个的酸酐醇盐并形成一个新的酯。一个切断键形成和切断键坏了。在这种情况下,羧酸盐是我们的离去基团。
第三个例子包括使用笨重的氢化物源di-isobutyl氢化铝(DIBAL- h)一个酯在较低的温度下形成一个醛。一个新的碳氢键的形成和切断键断裂。
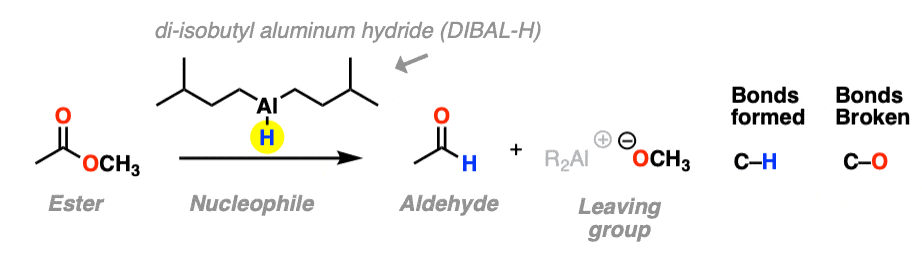
低温(-80°左右)这工作得很好;温度升高导致over-reduction。(请注意1]
3所示。Addition-Elimination机制与带负电荷的亲核试剂亲核酰取代
这些反应会如何工作的呢?他们不是年代N2型反应,因为年代N2只在sp是有效的3杂化碳原子。他们也没有确定年代N的一种反应离去基团第一,然后离开亲核试剂攻击(虽然这说明傅克酰化很好!)
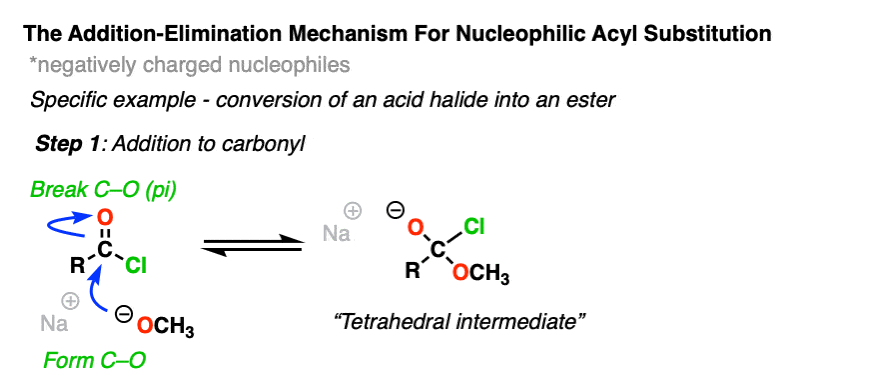
相反,两步实验提供强有力的证据addition-elimination通过一个机制四面体中间。(请注意2]
第一步应该非常熟悉的醛和酮的反应。(看到,简单的两步模式7键醛和酮的反应)
C = O组是碳亲电和反应容易与带负电亲核试剂通过一个除了机制(步骤1,形成C-Nu,打破切断(π)]
现在怎么办呢?通过添加的逆转机制,通过这个名字消除,四面体中间可以回复到起始原料,再生起始酰基组。
或者,如果我们有一个足够好离去基团,消除可能导致重新形成的切断和失去一个π键离去基团。
(步骤2,切断(π),打破C-LG]
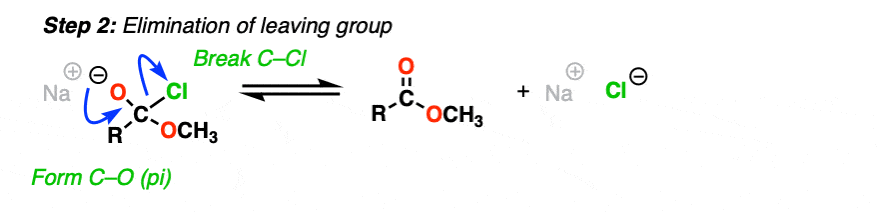
结果是,替换发生。
这两步机制被称为addition-elimination它的化学结构中是极为普遍的羧酸衍生品(即酰基组)。在这种情况下,一个中立的亲核试剂使用,或酸催化剂存在,一些干预可以延长机制(如质子转移步骤。P一个DPED),但仍addition-elimination核心过程。(想了解更多关于羰基消除——看Addition-Elimination)
4所示。亲核酰替换不工作
到目前为止还好,对吧?我们只需要添加亲核试剂酰基。的亲核试剂增加了,离去基团叶子,我们得到新的替代产品。简单!
这里有两个亲核酰取代反应的例子。
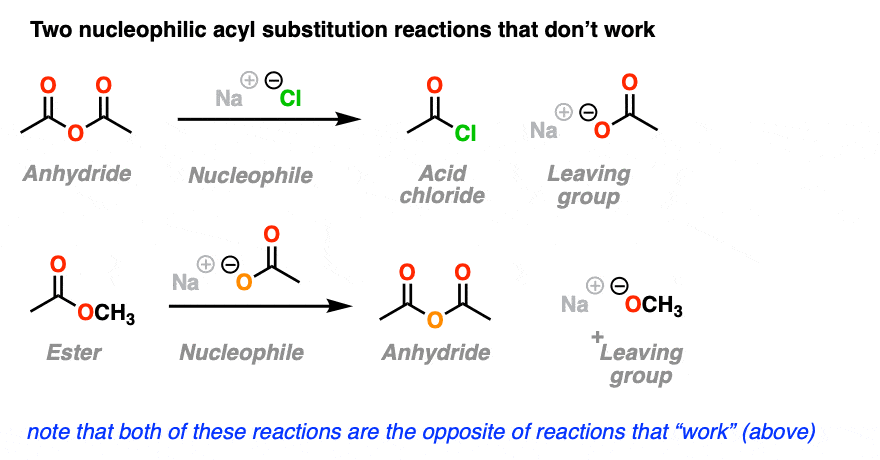
您可能已经注意到,这里显示的反应是亲核取代反应的完全相反的“工作”在上面的部分中,除了我们刚刚切换的身份亲核试剂和离去基团。
麻烦的是,尽管你可以画出这些“纸上”作为亲核酰取代反应,在现实世界中你可以等待大陆漂移泛大陆二世他们还不会发生。
为什么亲核酰取代喜欢向一个方向而不是其他?
5。NAS反应时青睐离去基团比亲核试剂较弱的基地
让我们来一个非常简短的旅行回到Org 1,我们学会了酸碱反应的黄金法则,也称为酸碱平庸的原则:最受欢迎的酸碱反应是一个方向更强的酸与一个反应强大的基础给一个较弱的酸和一个较弱的基础。(看到帖子:如何使用pKa表吗)
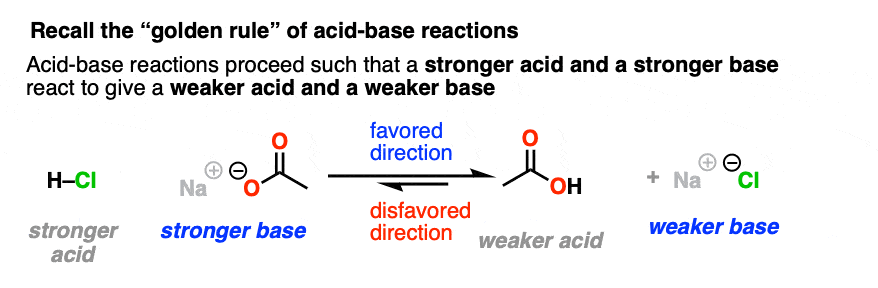
我们直觉地知道这一点,加入食盐水不会造成的云的盐酸气体和腐蚀性的氢氧化钠,而加入浓盐酸丸氢氧化钠不是一个清醒的人并没有很多的安全预防措施。
所以当试图考虑哪个方向是亲核酰青睐取代反应,预计它将有利于形成较弱的基础。
就像水往低处流,NAS反应将“流”,比如给更稳定阴离子(较弱的基础)。
(这也可能帮助认为“弱基地”的另一种方式说,“一对better-stabilized电子”)。
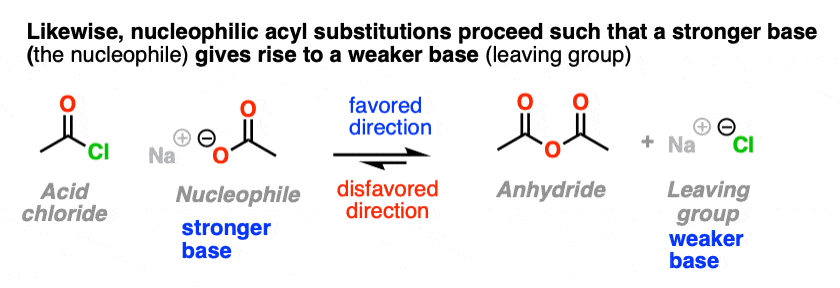
它还可以帮助我们理解为什么亲核酰取代反应不与醛和酮;它会导致氢化物离子(H)或负碳离子(R -),两者都是非常强大的基地。(注3]
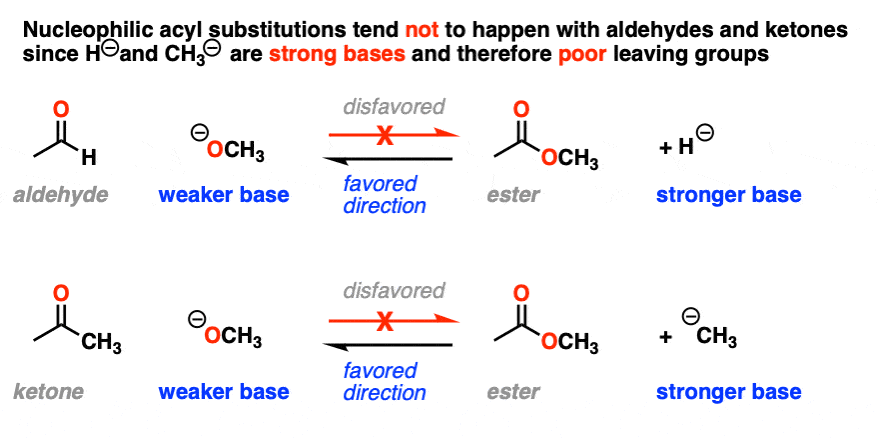
它可能帮助把这个图表的形式,如果我们图表pK一个(共轭酸)与亲核试剂/离去基团。
反应,涉及到一个“下坡“流亲核试剂来离去基团青睐,而那些需要一个较弱的“上坡”转换基地基地将走强不好的。
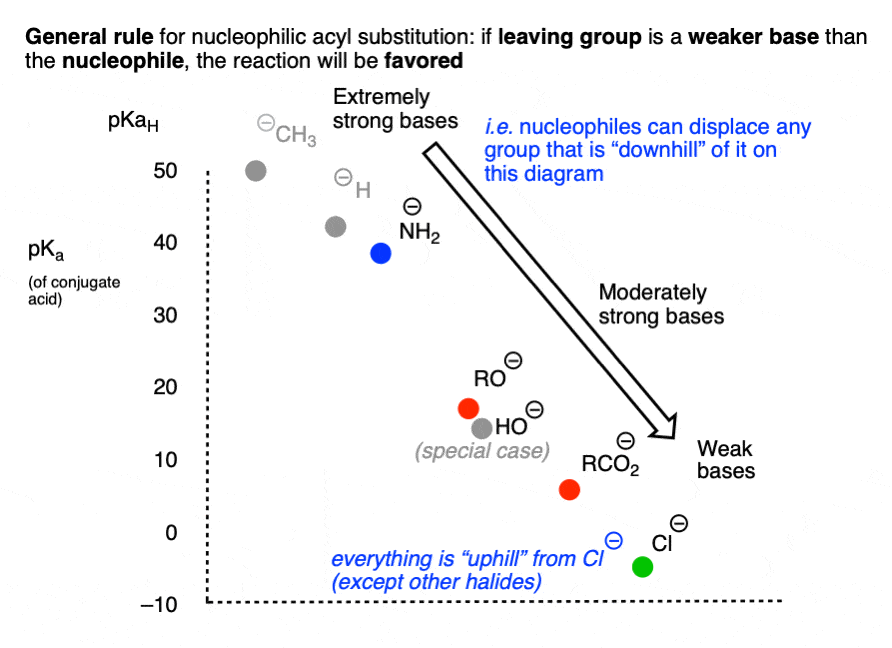
的另一种方法表达同样的想法是图如下,薄弱的基础(Cl)在哪里不能执行任何亲核酰取代反应,可能导致更强的基础:
原因很快就会变得清晰,HO(-)中省略了这个图表。(注意4]
徘徊在这个链接看到一个简化的反应能量图或点击这个链接。
{kind=link}
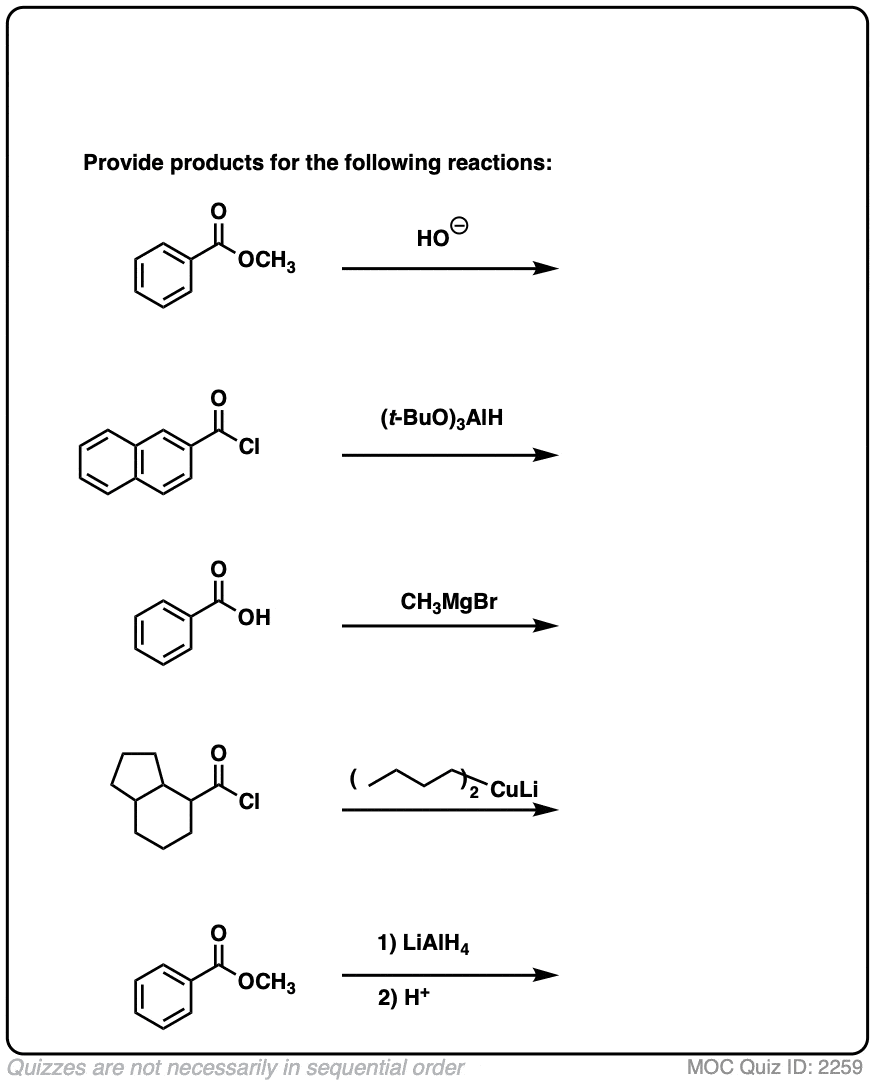
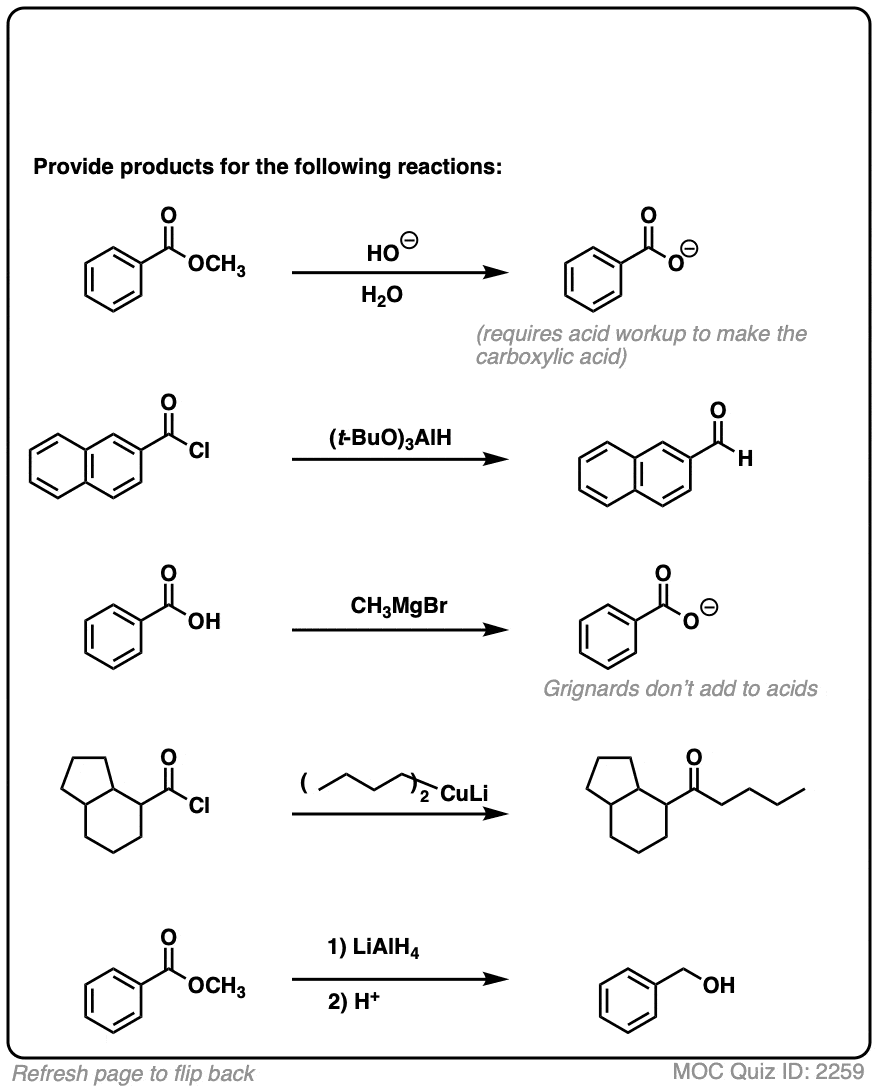
6。一些快速练习
让这个概念定居,为什么不测验自己一些快速练习?
首先,硫代酸酯。
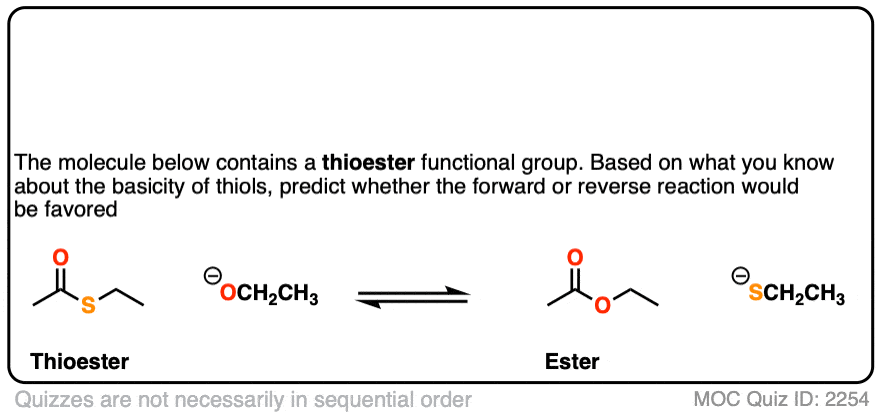 点击翻转
点击翻转
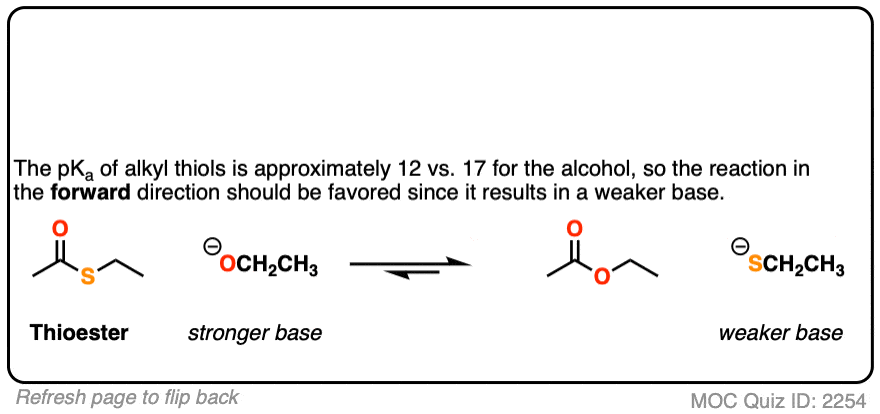
(注5]
第二,一个转换的一个例子酯到另一个酯产生一种被称为“酯交换”。
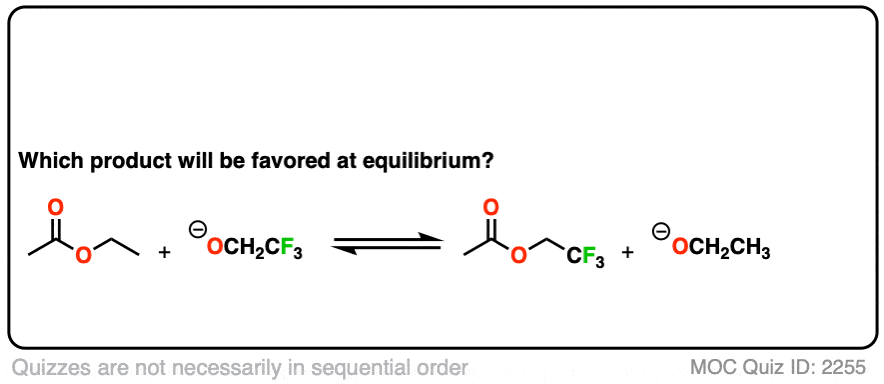 点击翻转
点击翻转
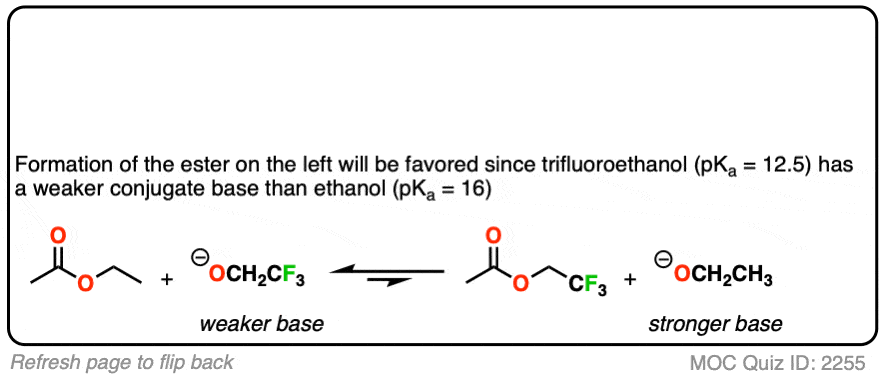
你可能会问,如果没有一个大pK一个之间的区别亲核试剂和离去基团吗?如果我们从乙酯例如,要转换成甲基酯吗?
自从pK一个的相似,方法之一就是“洪水区”亲核试剂(即使用大量过剩)驱动平衡对所需的产品。
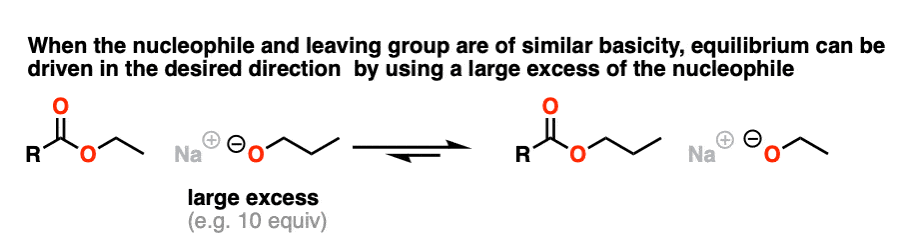
我不是说这是最好的(转换成卤酸或酸酐接着用一种酒精将上级),但它仍然是可行的。
7所示。羧酸…。酸!
现在,我们有一个处理的关键因素影响亲核酰替换,让我们来看一个例子,应该看起来简单…但不是。
氢氧化钠是一共轭碱水(pK一个14)和NaOCH3是共轭碱CH的3哦(pK一个16)。
所以我们知道,反应产品应该有点忙. .对吧?
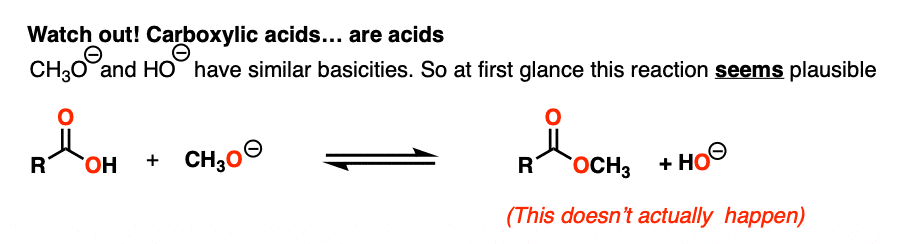
实际上…不!
这是因为有一个额外的因素,我们需要考虑。羧酸是酸和酸碱反应快。(见:酸碱反应快)
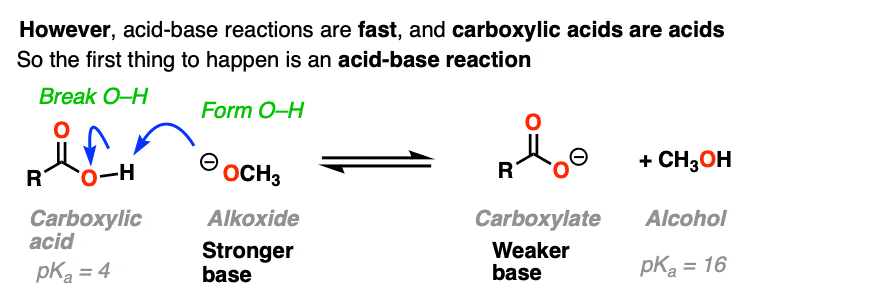
如果亲核酰取代发生在这一点上,它将不得不增加羧酸盐这将使一个di-anion两个负电荷在同一分子(如果这听起来不稳定——这是! !)。
这将是O (2 -)!
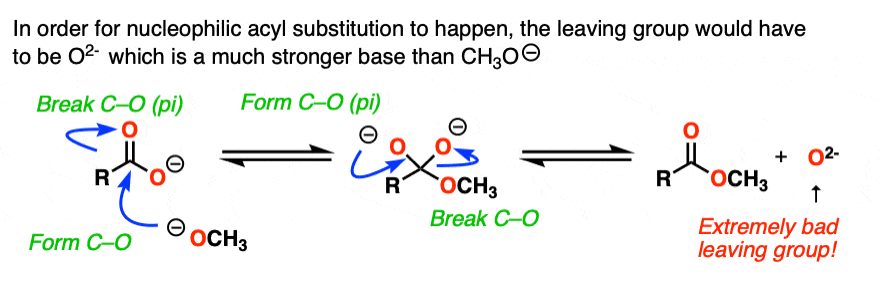
有一个例外注6),这不会发生。
请注意,羧酸可以被转换成酯类,但需要酸(如反应费歇尔酯化)。在本文中,我们关注带负电的亲核试剂。
8。皂化酯的基础
如果添加RO (-)羧酸结果在去质子化,相反:添加的氢氧根离子酯吗?
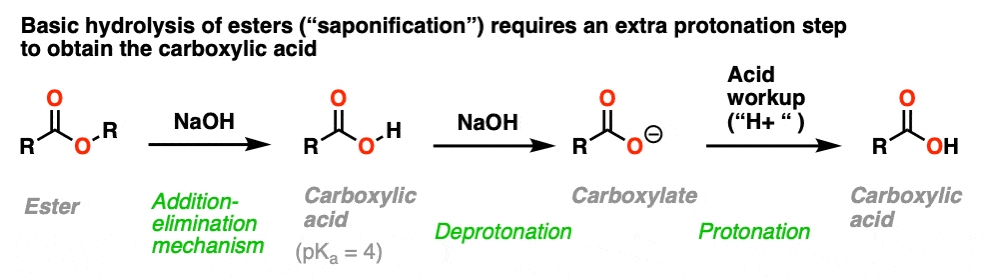
这是一个众所周知的过程称为皂化(或者只是“基本水解”),非常好用。
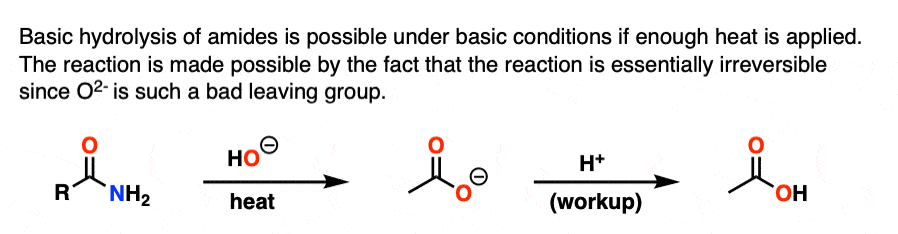
替换部分的反应通过熟悉的收益除了- - - - - -消除的过程。这个结果在一个羧酸。然而,由于羧酸是酸的,和基本条件下发生反应,生成的酸会迅速被氧化deprotonated给羧酸盐。
因此获得中性羧酸结束时的反应,必须添加酸在检查步骤。
“皂化”这个名字来自于经典中使用这个反应肥皂(长链脂肪酸的羧化物)治疗脂肪(酯类长链脂肪酸的甘油)碱液碱金属氢氧化物(一个通用的术语)。
9。分子内NAS
它总是有用的思考你学习任何新反应的分子内变异。亲核酰替换也不例外!
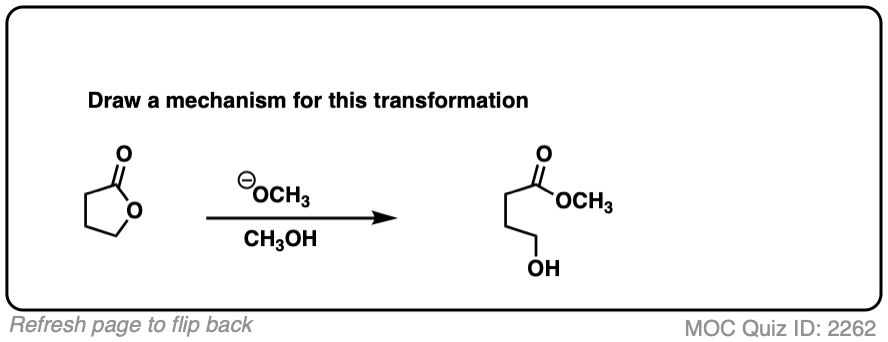
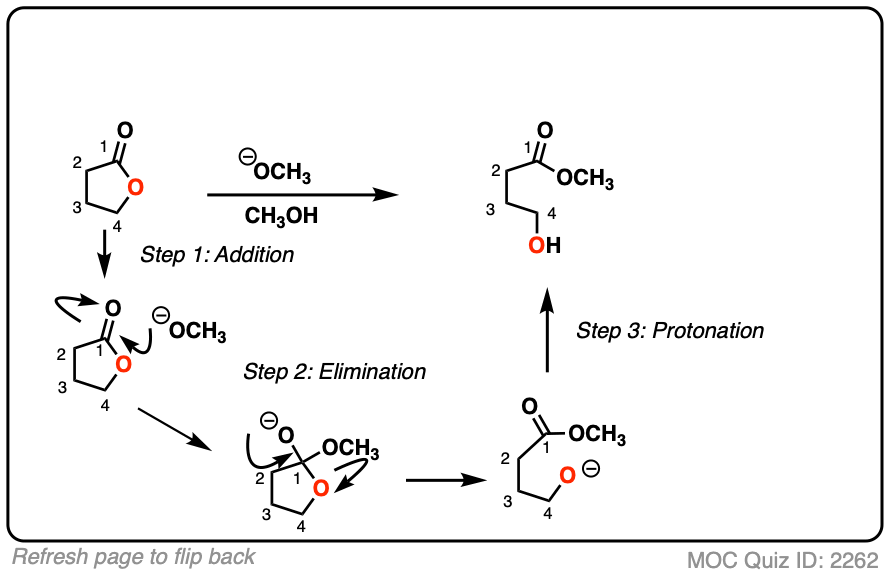
如果一个五或六元环可以通过亲核酰取代形成,它通常会。在这个例子中,治疗与强碱氢化钠(不)的结果醇盐形成一个新的戒指,甲氧基取代(CH3O (-))
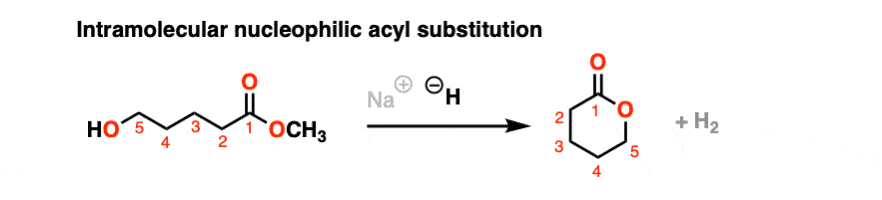
徘徊在这个链接看到的机制或点击这个链接。
{kind=link}
这是一个有趣的例子。这里正在发生什么?
 点击翻转
点击翻转
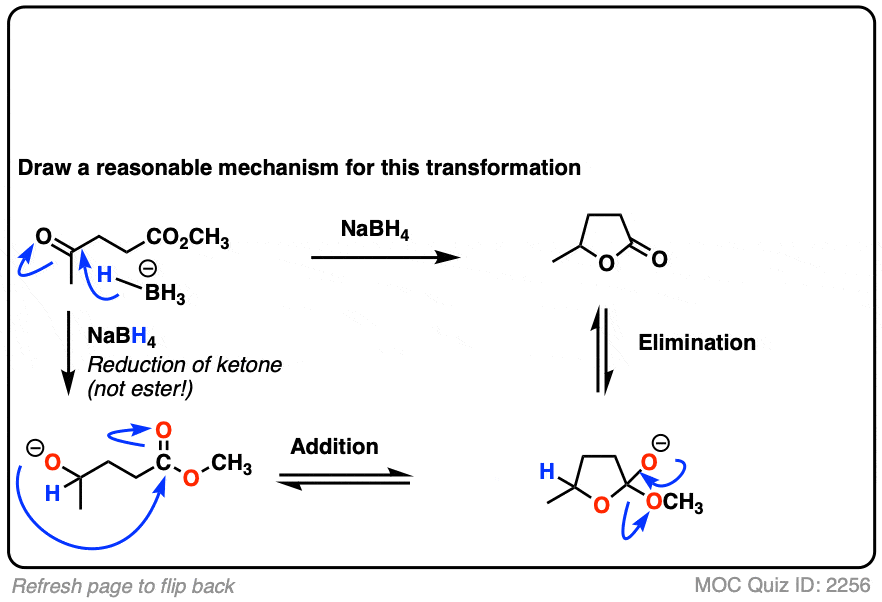
(提示:去质子化并不是唯一的一个醇盐)
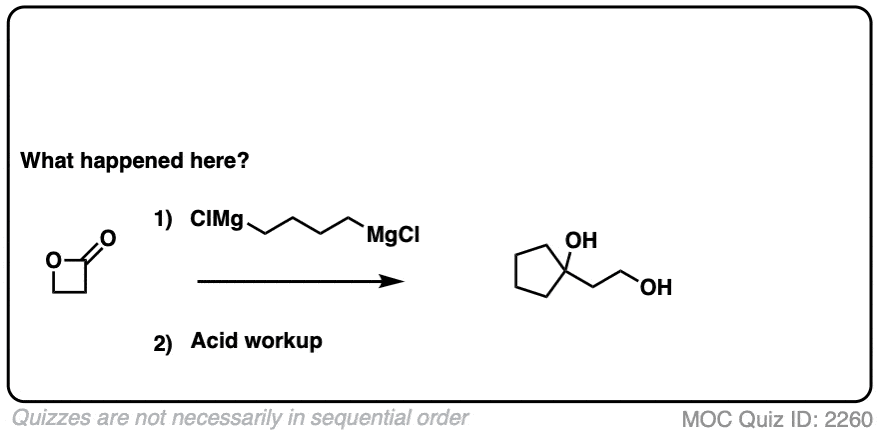
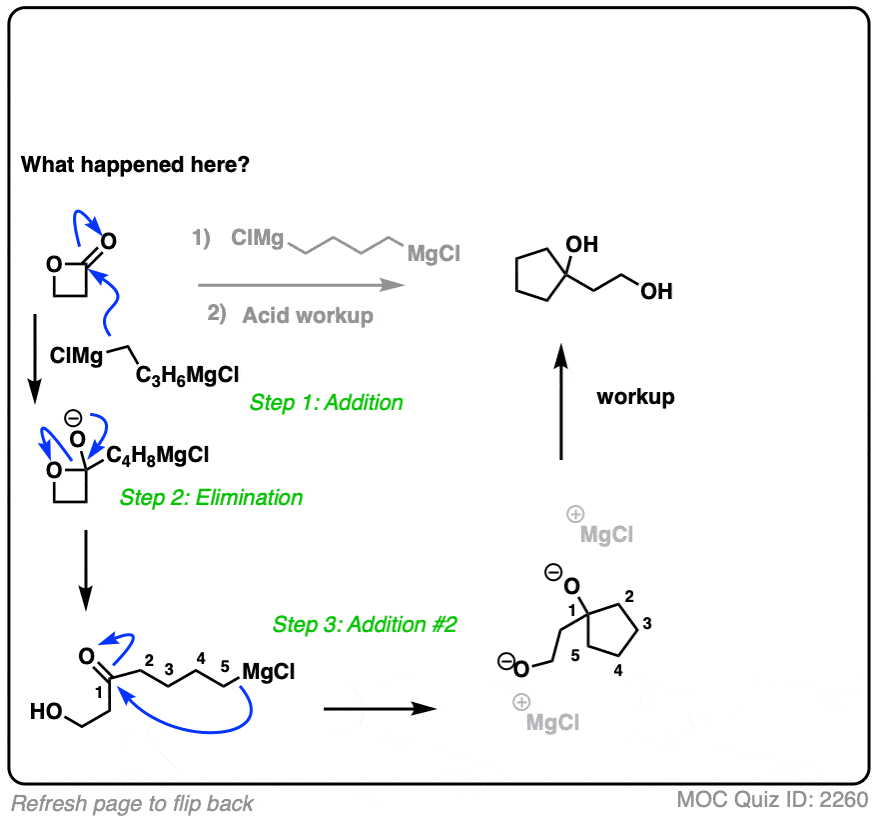
10。——格里纳和LiAlH当亲核试剂添加两次4
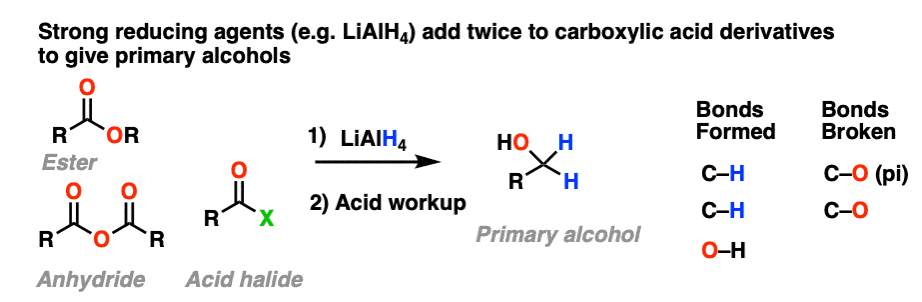
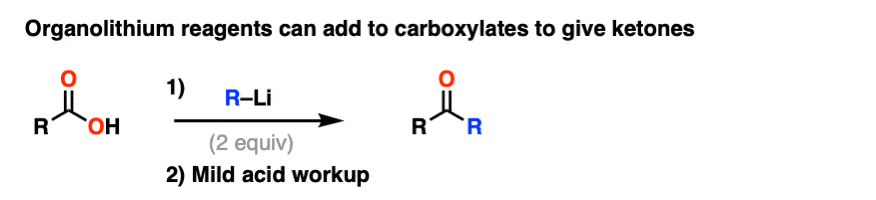
格氏试剂和强大减少代理像LiAlH4倾向于增加羧酸衍生品两次。
格氏试剂(organolithium试剂)叔醇从酯类、酸卤化物和酐。
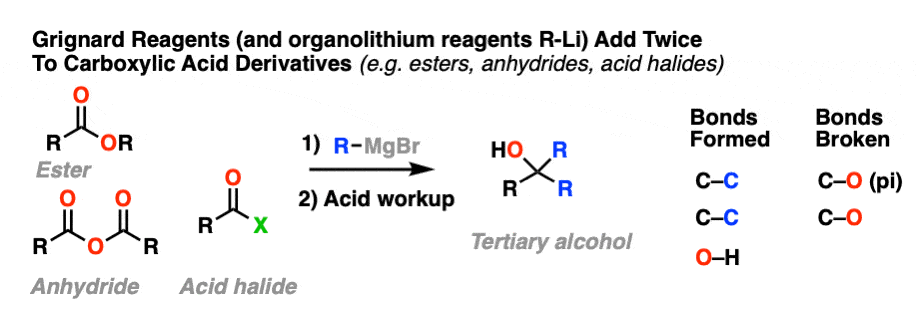
强大的减少代理(例如LiAlH4)给主要醇从酯类、酸卤化物和酐。
所以这些“双加法”反应如何工作?
除了的亲核试剂R(-)或H(-)紧随其后消除的离去基团最初给酮(格氏)或一个醛(LiAlH4)。
如果消除过程快相对增加,那么醛或酮会形成强大的存在亲核试剂之前所有的开始酯(或其他羧酸导数)消耗。
由于酮(醛)亲电试剂比酯类(即。他们用的亲核试剂反应更快),结果,有时我亲切地叫饼干怪兽反应:反应形成产品更多的反应向亲核试剂起始物料。
(因为饼干怪兽不能停止后只有一个Cookie)。
这个结果除了成为一个反应发生酒精后的酸。(即使一个相当于格氏使用,该产品仍然是一个三级酒精和未反应的酯。)
所以最终被最后一个序列除了- - - - - -消除- - - - - -除了其次是最终检查(质子化作用)。
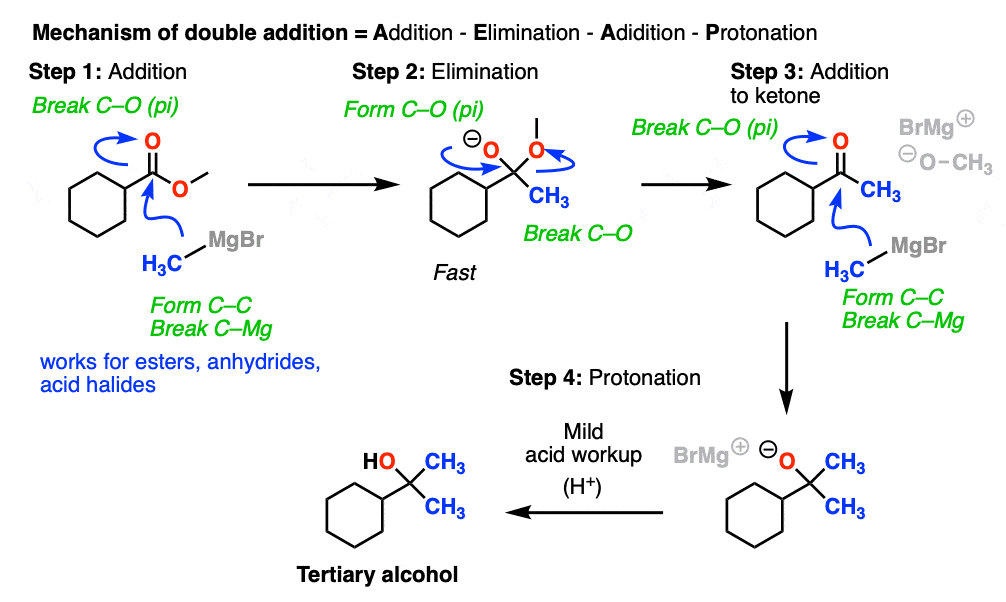
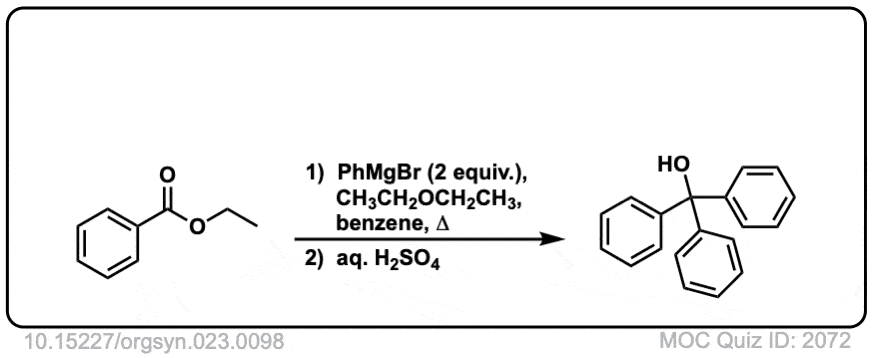
这是一个具体的例子:
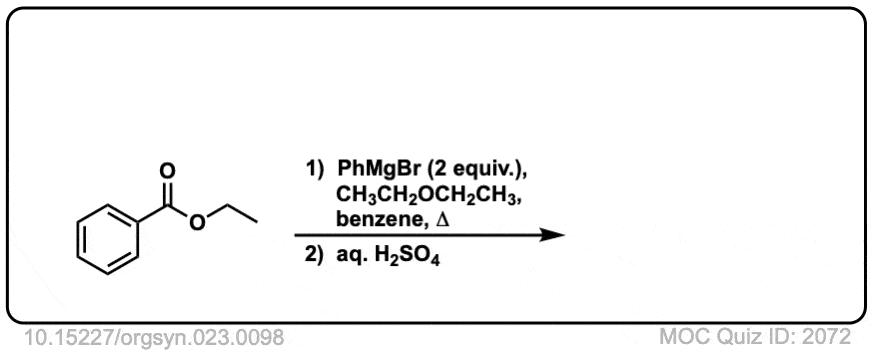 点击翻转
点击翻转
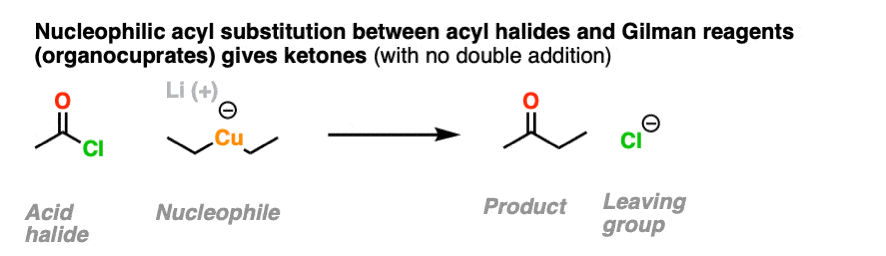
可以得到除了酸卤化物停止的酮阶段通过使用organocuprates(吉尔曼试剂)不那么贪婪的格氏或organolithium试剂。
11。中性的亲核试剂和酸催化
这整篇文章一直局限于的反应消极的带电的亲核试剂,如格氏、氢化物、酰胺基、醇盐,羧化物和卤化物。
我们已经看到,在这些条件下,平衡的方向几乎完全是由稳定的离去基团。这使得它很难执行替换酰胺的反应,例如,因为R2N(-)是这样一个强大的基础。
有另一种方式吗?
是的。我们应该期望增加酸将协助执行亲核酰替换,因为共轭酸总是更好的离去基团。
这的确是事实。看到Addition-Elimination反应与中性的亲核试剂(包括酸催化)
这里的权衡是酸性条件不兼容极其基本的亲核试剂如格氏和氢化试剂。
我们将探讨亲核酰取代在中性和酸性条件下在下一篇文章中。
12。总结
与带负电的亲核试剂亲核酰取代反应所得通过addition-elimination机制。
- 在第一步(除了)亲核试剂攻击羰基碳,导致四面体中间。
- 在第二步中(消除)碳-氧生成和π键离去基团是流离失所。
- 因为我们正在形成,打破债券在同一个碳这是归类为取代反应。
- 向前和向后的反应可以被视为一个平衡。像一个酸碱反应,平衡将有利于生产的方向薄弱的基础。
- 羧酸在基本条件下一般不进行亲核取代,因为他们是如此容易deprotonated和由此产生的离去基团(O2)是非常基本的。
- 然而,酯类进行基本的水解与氢氧根离子(“皂化”)给羧酸(质子化作用后)。
- 分子内的亲核酰取代反应可能发生,特别是当五年和六元环可以形成。
笔记
【注:这篇文章明显从2011年更新一篇文章,题目是“简化的反应羧酸反应,第1部分)。
注意1 -技术上的第一步DIBAL减少- h是协调的羰基氧气Lewis-acidic铝,紧随其后的是交付的氢化物。但是对于我们的目的,我们可以看看它的H (-)羰基碳。
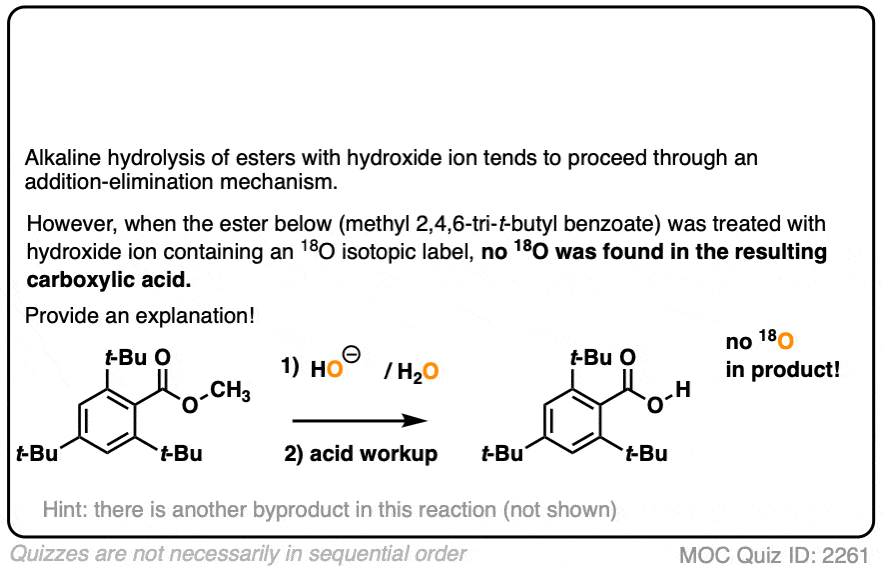
在第一个实验中(裁判),一个酯是包含isotopically贴上氧氢氧化处理(18与“正常”的阿16O)。没有恢复酒精包含了同位素标签。
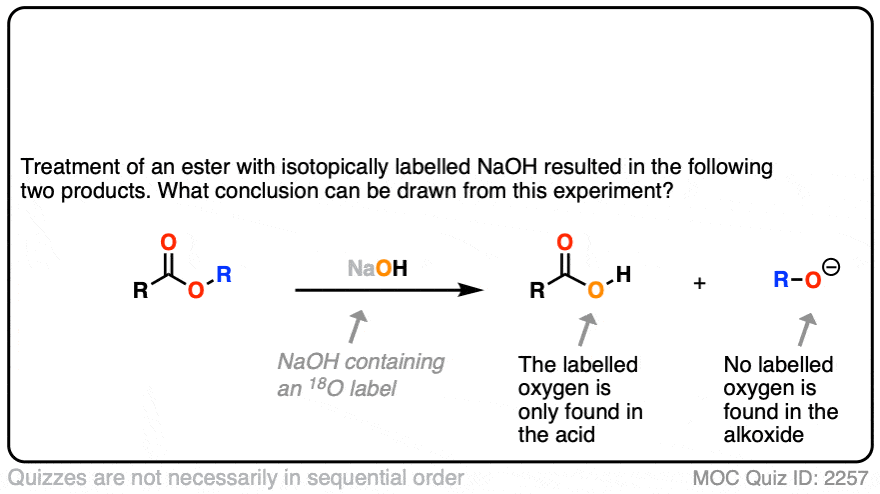 点击翻转
点击翻转
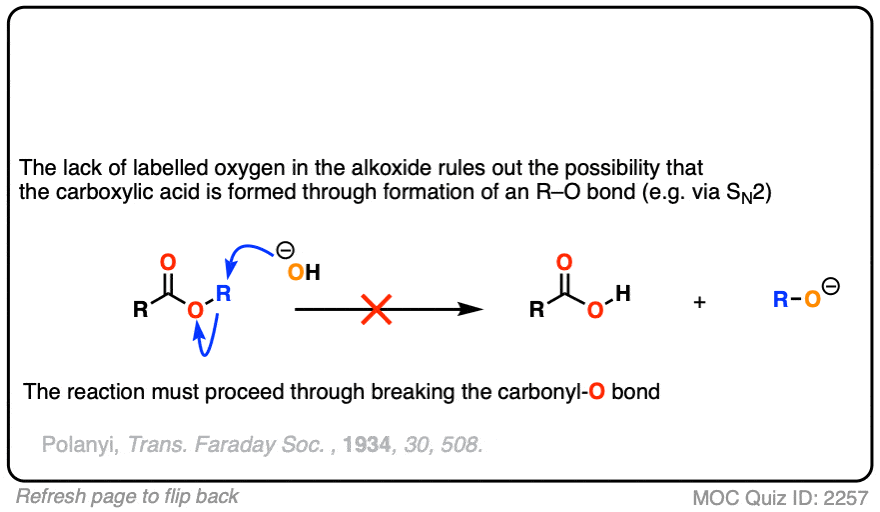
结论是羰基。氧气键坏了,不是烷基。氧气债券(如在某种形式的年代N2)过程。类似的实验已经完成了酯类与手性醇,水解醇被发现维护他们光学纯度。
在另一个经典的实验(裁判)一个酯是处理18O标记氢氧根和反应是停止之前完成。
复苏开始酯显示一些公司的18在阿羰基氧气。这结论可以从什么?
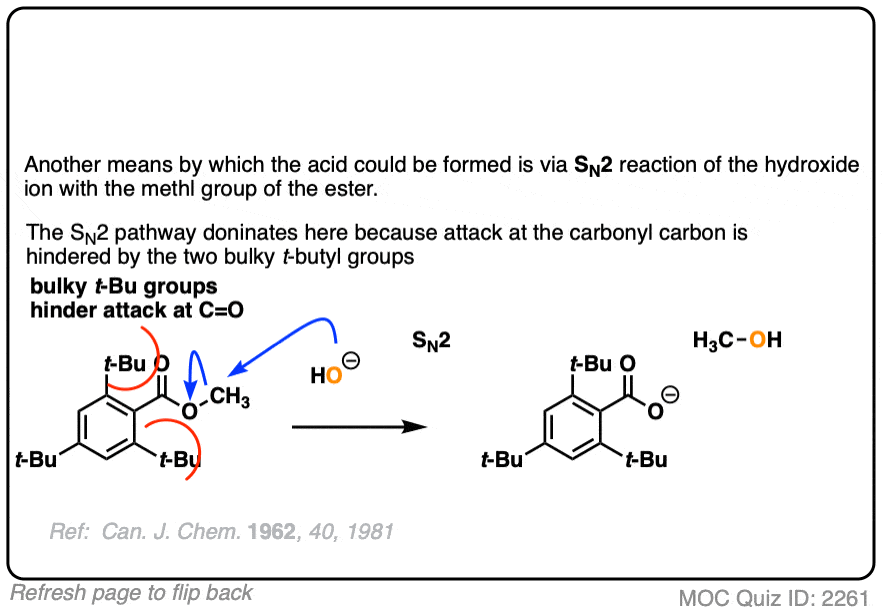
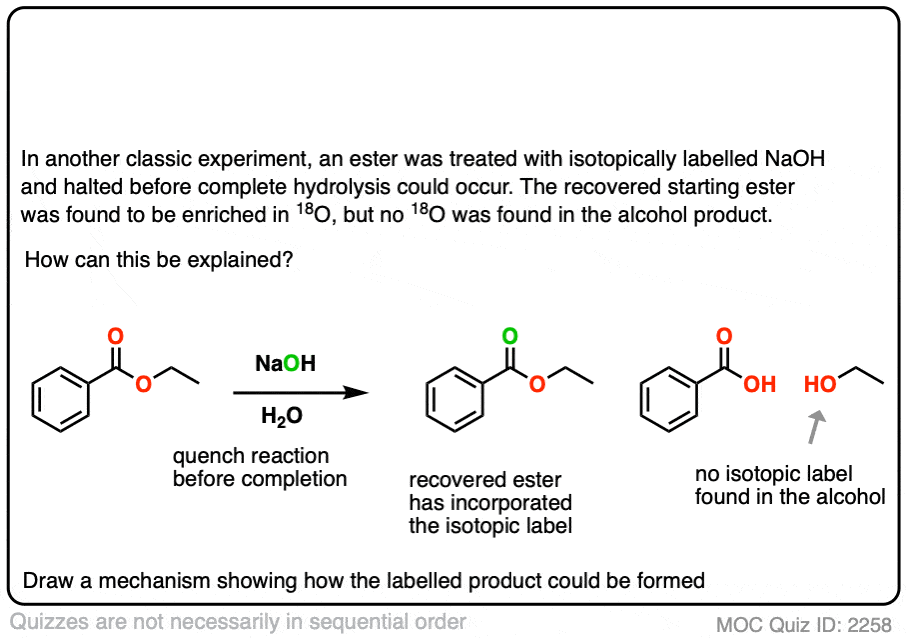 点击翻转
点击翻转
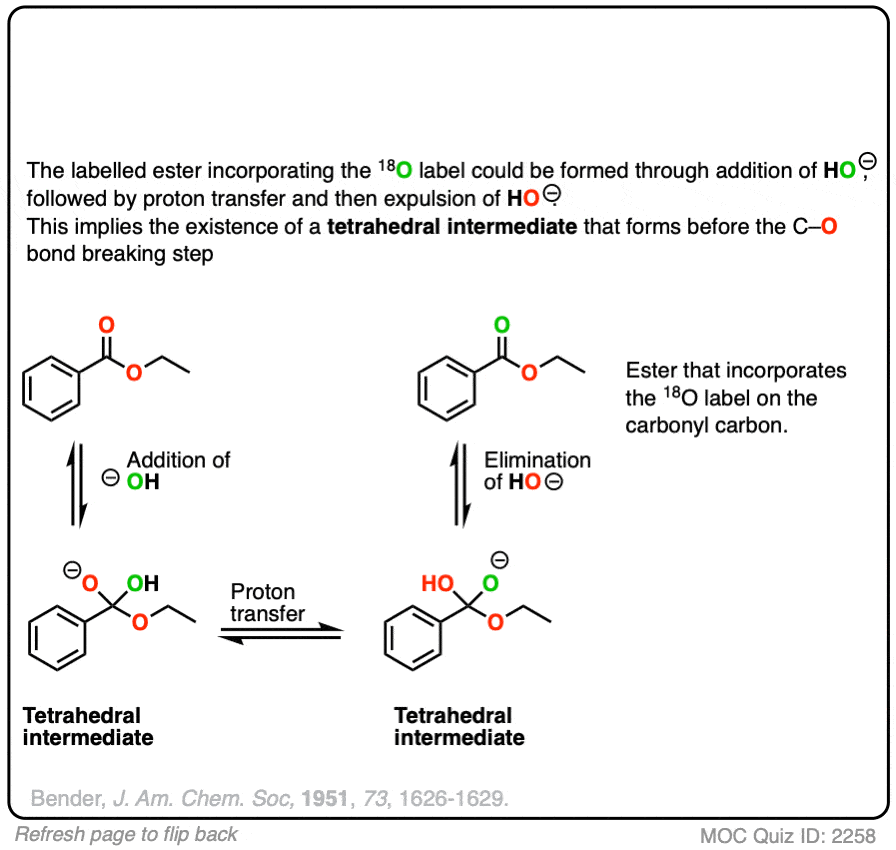
可逆的18O标记HO(-)导致了四面体中间。假设18O和16O标记本质上相同的反应性氧(他们这样做,在几个百分点其次是消除),质子转移16哦会导致一个开始酯富含18o .交换或者组织的不应该发生。
这是一个关键的实验,建立了四面体中间的存在。
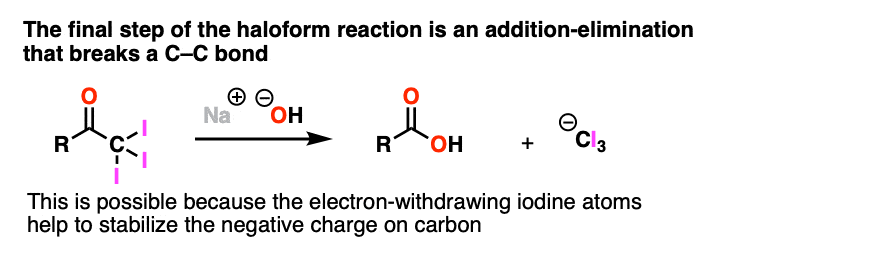
注3。一个明显的例外是最后一步的卤仿反应(见:卤仿反应)addition-elimination与氢氧化钠亲核试剂结果(-)CI的驱逐3作为一个离去基团。这只因为吸电子卤素有助于稳定产生的负碳离子。
注意4。有足够的热量,就可以执行基本的酰胺水解,外表的NH以来被“上山”2(-)比HO(-)更强的基础。然而,一旦羧酸形成,去质子化的结果吗羧酸盐从位移反应基本上呈现不可逆转的北半球2(-)会给O (2 -)
注5。自然广泛采用亲核酰取代反应、硫代酸酯是重要的“酰基转移”在生物化学试剂。硫的辅酶A作为离去基团与各种亲核酰取代反应。
特别重要的是甲羟戊酸途径导致的生物合成萜烯和类固醇。
注6。尽管格氏试剂只会deprotonate羧酸给羧化物,organolithium试剂将增加羧化物给稳定的四面体中间,分解酮在酸性表面的污迹。
测试你自己!
 点击翻转
点击翻转
 点击翻转
点击翻转
 点击翻转
点击翻转
 点击翻转
点击翻转
(高级)引用和进一步阅读
3月份的先进的有机化学,凯里&桑德博格部分(第三章)和B(第七章)是好先进的关于这个主题的来源。bdapp平台有机化学结构和机制由c·k·英格尔德的历史背景。
1。在水解的机理。醋酸戊酯的碱性皂化
m .波兰尼和a . l .萨博。
反式。法拉第Soc。1934年,30岁,508 - 512
DOI10.1039 / TF9343000508
早期研究酰基取代的机制使用isotopically贴上氧气识别acyl-O债券裂缝的主要机制酯水解。
2。氧气交换作为一个中间存在的证据酯水解
Myron l·本德
美国化学学会杂志》上1951年73年(4),1626 - 1629
DOI:10.1021 / ja01148a063
经典的实验建立四面体中间的存在在亲核酰替换。本德苯甲酸乙酯的偏酸性水解18O浓缩水和恢复18O-enriched苯甲酸乙酯。
3所示。立体化学的影响strains-XIII:硼氢化钠的反应动力学羰基团体的方便工具调查醛和酮的反应活性
H.C.布朗,平炉惠勒,k .川
四面体,1(3),1957年,214 - 220。
DOI:10.1016 / 0040 - 4020 (57)88041 - 7
有用的研究增加的速率常数NaBH4不同的醛和酮。
4所示。亲核的反应的催化机制羧酸衍生品。
Myron l·本德
化学评论1960年60(1),53 - 113
DOI:10.1021 / cr60203a005
审查机制的亲核酰替换。
非常感谢你詹姆斯。它确实是有帮助的。
我希望更多的为你特别是你的研究进展
你好詹姆斯,谢谢你的网络。我注意到DIBAL-H在你画一个碳原子。
问候,
鲁本
为什么相应的羧酸酸性的增加,由于附件苯或乙烯基群,相反减少预期由于共振效应?
好问题。很可能由于sp2杂化碳原子的诱导效应的芳环羰基。你可以把它们有效的电负性更大,因为他们有33%提到(25% vs sp3)和电子和核的关系更加紧密。
酯的亲核酰取代的哦,为什么还是——集团离去集团?
图中显示的pKa离开团体,看来,哦,是一个稍微比或者——更好的离去基团。如果是这样,为什么反应还出现吗?图的状态,这是一个特例,但我真的不知道为什么。
好问题。亲核酰取代的产物是一种羧酸。
现在你有一个羧酸(pKa = 4)醇盐的存在(共轭碱的酒精,pKa约16)。
然后deprotonated羧酸盐。
现在发生了什么?
如果您添加RO羧酸盐(-),为了让亲核酰替换发生,你的离去基团必须O (2 -)。基本上,这是不可能的。
精心设计!这种狂热的学习者的宝石:)
谢谢你!
”4。亲核酰替换不工作”
标签的酯混合酸酐。
我真的很喜欢你的网站,谢谢!:)
谢谢你的小心!固定的。
上帝保佑你! !非凡的工作这样一个困难的课程! !
你的网站是如此珍贵。它帮助我爱上有机化学和ace我考试。非常感谢。
嗨,玛丽,谢谢你,我很高兴你找到该网站有帮助。最好的祝愿,詹姆斯
真正伟大的表示。真正有用的高级竞争考试。
亲爱的先生,
真的帮了我很多在我的化学IIT JEE,我做准备。
~ Abhay