烯醇、烯醇化物
烯醇化物——形成、稳定性和简单的反应
最后更新:2023年5月25日|
所有关于烯醇化物
- 烯醇化物可以通过去除形成质子在碳原子相邻吗羰基(即“α碳”)。由此产生的阴离子比典型的烷烃更稳定自负电荷可以通过共振离域的氧原子。
- 从醛和酮烯醇化物可以形成,但也从酯类酰胺,腈和许多其他羰基包含的化合物。当一个额外的吸电子集团存在的α碳烯醇化物变得更容易。
- 烯醇化物是的亲核试剂和有很多有用的与亲电试剂反应。在这篇文章中我们将介绍与H +反应的烯醇化物,卤素,简单的亲电试剂。
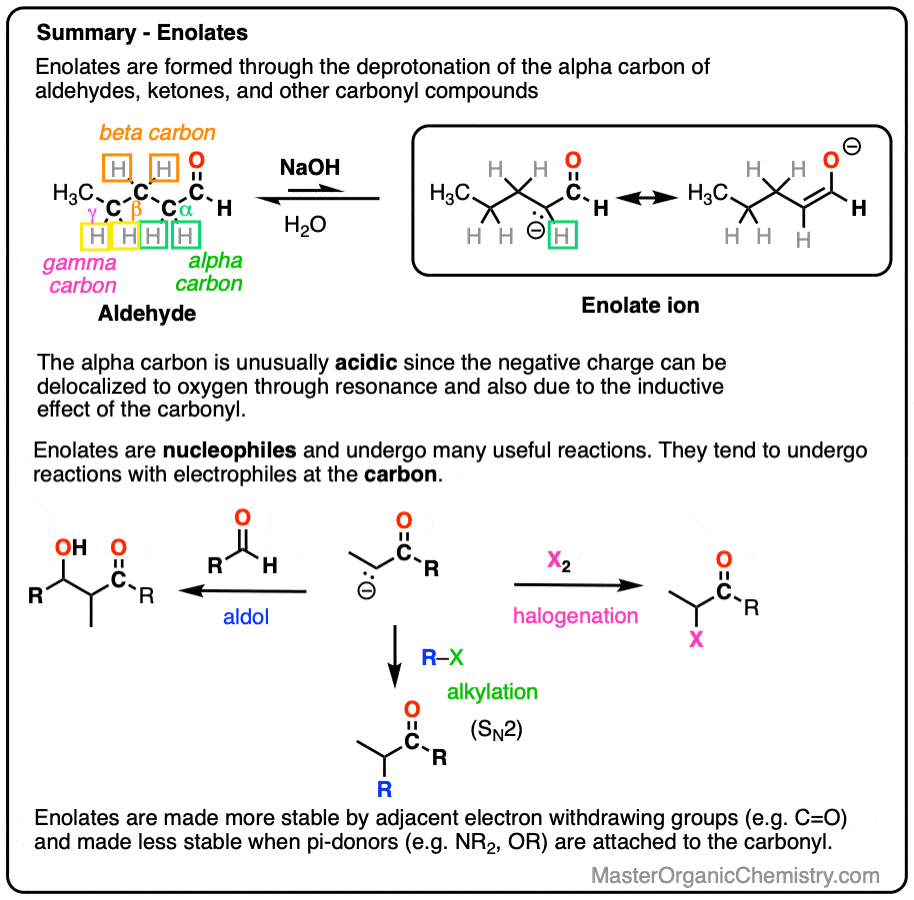
表的内容
- 烯醇化物离子是什么?
- 通过烯醇化物Keto-Enol互变现象
- 烯醇化物往往与亲电试剂反应的碳原子
- 差向异构化作用通过烯醇化物(含重氢)
- 烯醇化物是良好的亲核试剂!一些简单的例子(卤化,醇醛)
- 影响烯醇化物稳定性的因素
- 烯醇化物酯和酰胺
- 酮烯醇化物
- 结论
- 笔记
- 测试你自己!
- (高级)引用和进一步阅读
1。烯醇化物离子是什么?
醛和酮的反应与强碱(如。HO(-))给一个烯醇化物离子。
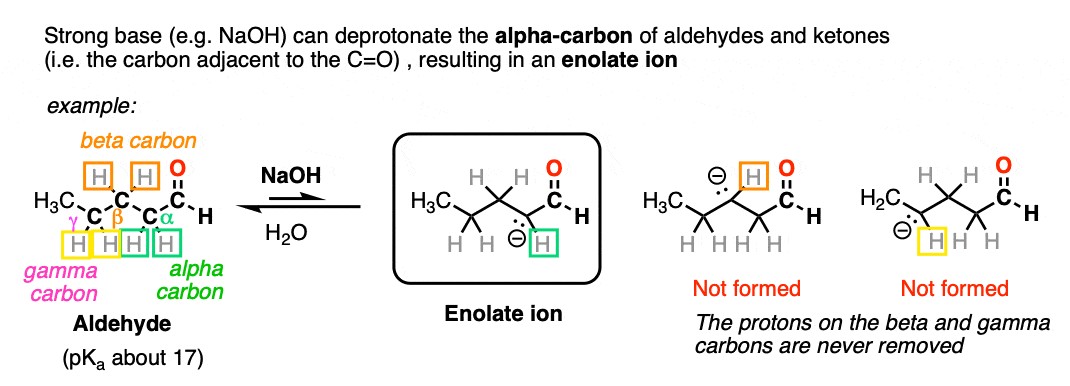
碳的碳氢键坏了相邻到羰基碳(即“α碳”)。碳进一步远离羰基永远不会deprotonated。
反应是可逆的,平衡倾向于起始原料约100:1 [注1这仍然是一个相当惊人的结果,我们知道碳氢键。毕竟,烷烃非常耐基地(pK一个这里的东西是让= 50)醛或酮大约30多pK一个单位(= 1030.)比通常是酸性的。
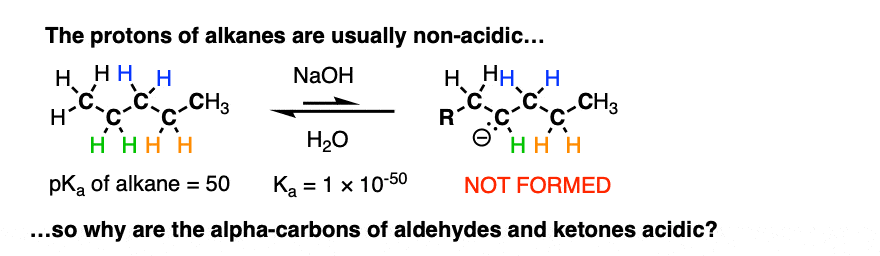
这是为什么呢?
要理解酸度,是首先要考虑的问题检查的稳定性共轭碱。在组织我们看起来不同影响酸度的因素并指出负电荷是稳定的
- 归纳影响(邻吸电子组)
- 共振移位(特别是负电荷可以放在一个电负性原子)
- 增加了电负性的原子(F > O > N > C)轴承的负电荷
(看看所有这些概念我们学习有机化学第一季度继续相关的许多章节之后!)
如果我们比较共轭碱烷烃与共轭碱一个醛或酮,有两件事应该突出。
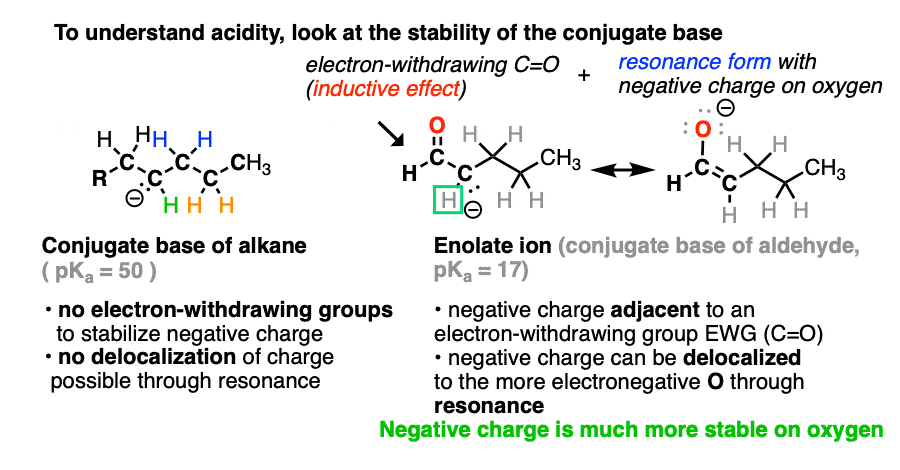
请注意,如果我们表单上的负电荷β——或者γ——碳、共振非局部化是不可能的。
为了一个烯醇化物形式,必须上一个氢α碳。如果一个醛或酮缺少一个质子α碳我们称之为“非enolizable”。
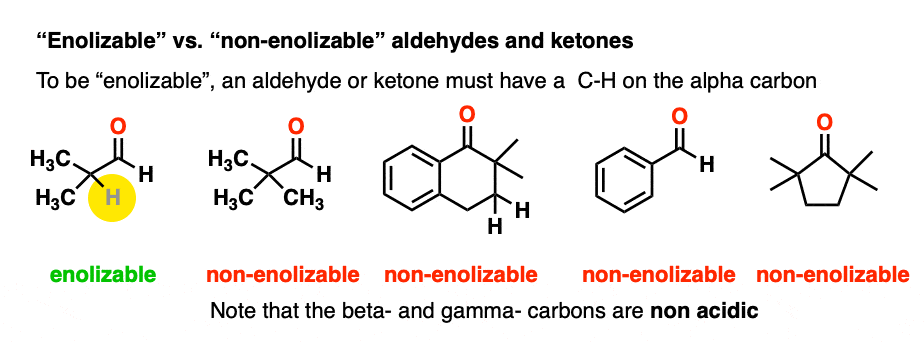
事实证明,烯醇化物离子是非常重要的中间体对许多反应,我们会花大量的时间去了解他们。
我们首先覆盖他们的一些简单的反应,然后完成通过探索各种影响其稳定性的因素(反应)。
2。通过烯醇化物Keto-Enol互变现象
烯醇化物可以被认为是醛和酮的共轭基地(等等),但他们也的共轭基地烯醇。
酮烯醇互变现象可以通过加入酸或碱的醛或酮。如果你需要复习一下,回去看看这篇文章:Keto-Enol互变现象]
一般来说的酮互变异构体被看好在平衡最主要的原因在于切断π键约20千卡每摩尔比碳碳π键]。
当酮烯醇通过增加基础发生互变现象,它通过一个中间的形成烯醇化物使质子化和转换烯醇的形式。
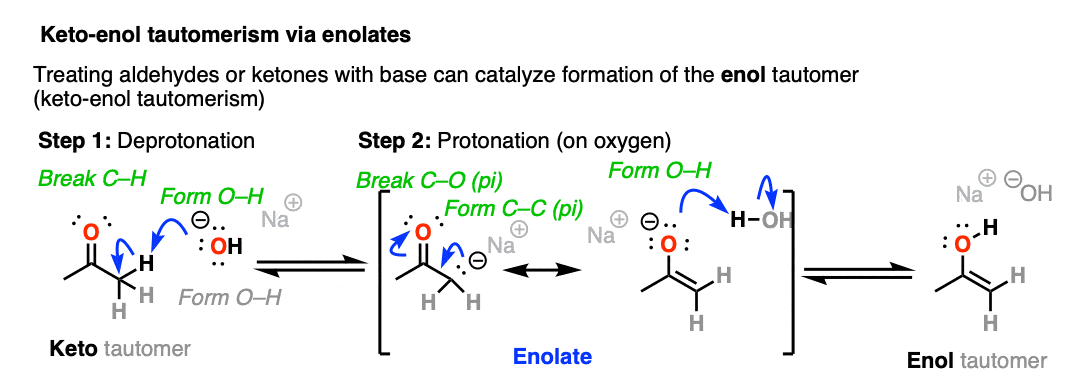
自醛/酮(pK一个比H = 17日至19日)是一个较弱的酸2O (pK一个15)平衡是主要的左,这意味着烯醇化物是一种微量组分的反应混合物。
你会原谅如果这似乎并不很令人兴奋。
这一切发生的基础是被用来形成一个复合的烯醇),这是在大多数情况下数量至少100:1平衡。谁在乎呢?
令人兴奋的是烯醇化物本身就是一个伟大的亲核试剂,与其他亲电试剂反应,如果他们碰巧在场,如:
我们会得到这些!但首先,一些后果烯醇化物形成。
3所示。烯醇化物往往与亲电试剂反应的碳原子
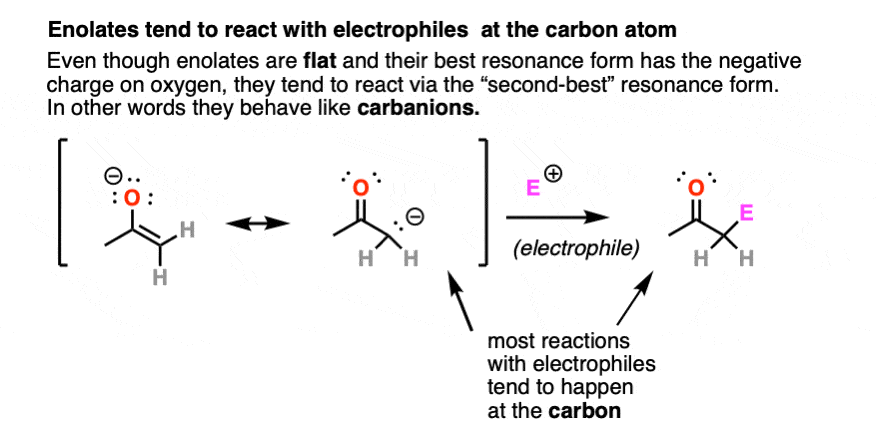
烯醇化物有两个突出的共振形式,可以画一个负电荷碳或一个负电荷氧气。
他们是平,所以他们最“正确”的形式是吸引他们的负电荷氧气。
然而,他们往往就像负碳离子反应,通过碳原子最亲电试剂进攻。有办法得到氧气更被动,而不是碳,但现在我们将推迟这个问题更高级的课程(注3]。出于我们的目的在这里,我们将用碳上的负电荷。
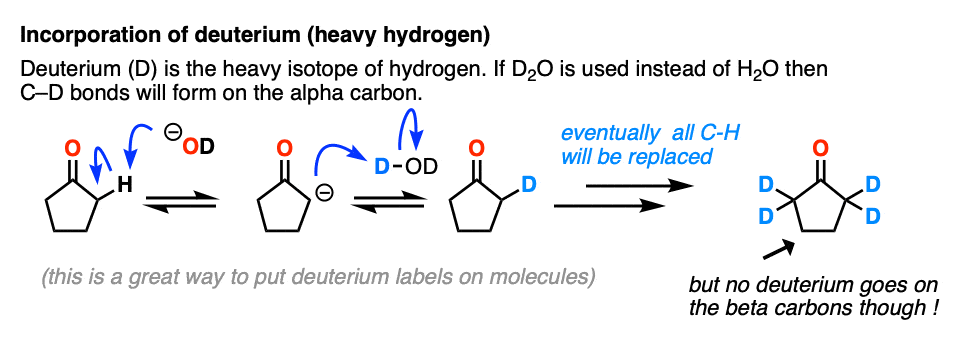
4所示。差向异构化作用和含重氢的烯醇化物
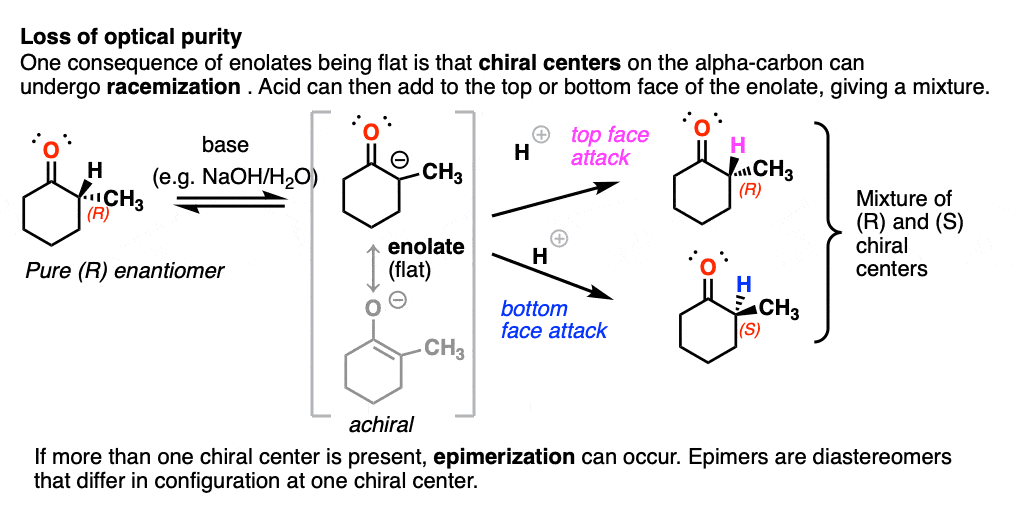
烯醇化物的平面度的一个重要后果。
如果你开始与一个手性中心α碳和治疗基地,α碳持平,失去其手性。如果烯醇化物然后用另一个反应亲电试剂(如H +),它可以从扁平的脸烯醇化物,导致外消旋化碳。(更具体地说,我们通常称这个过程差向异构化作用(注意4因为它不会导致一个外消旋混合物的对映体如果另一个手性中心。
(手性中心的β-或γ-碳不是影响因为这些碳氢键不是酸性)。
只是为了好玩,如果我们使用氘溶剂和维2O代替H2O(回想一下,D是氢的重同位素)然后我们将取代碳氢键和c - D债券。这可能是一个有用的结合方式氘。
5。烯醇化物的亲核试剂是
卤化
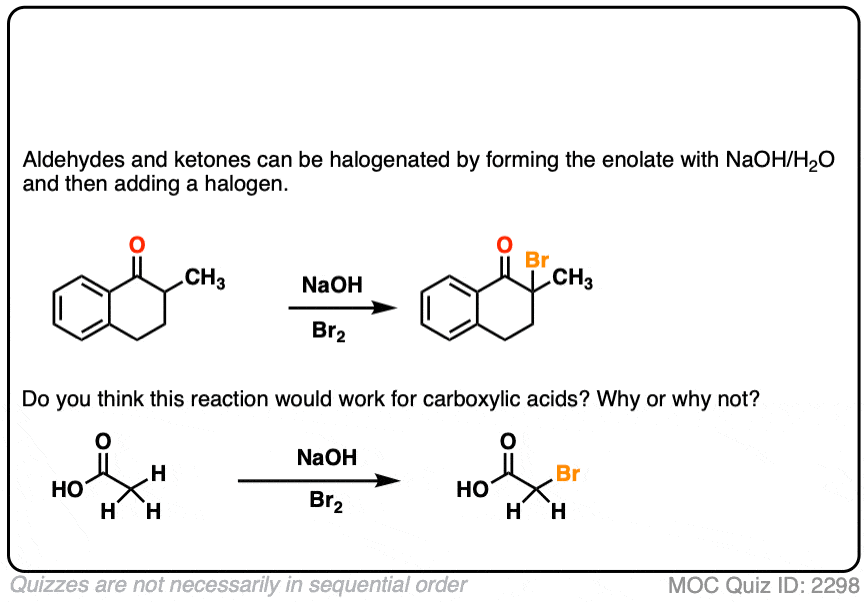
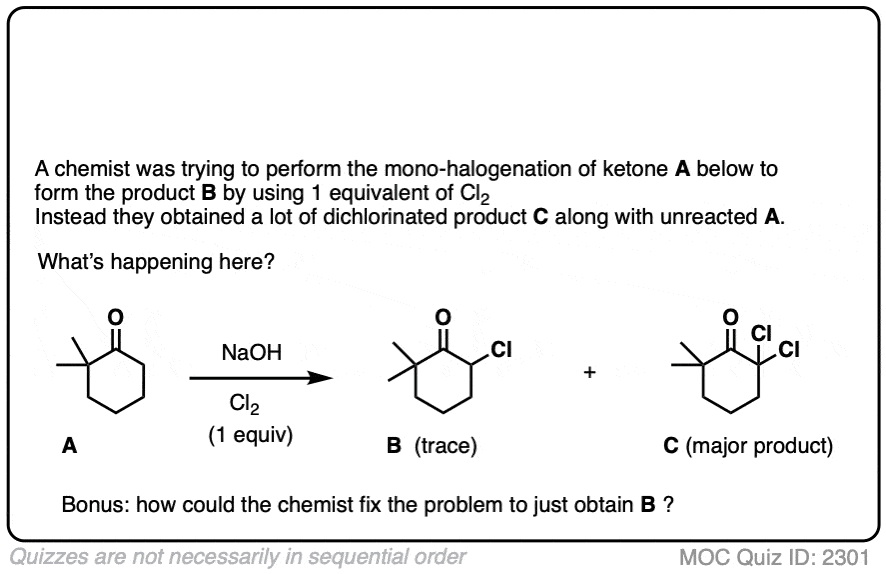
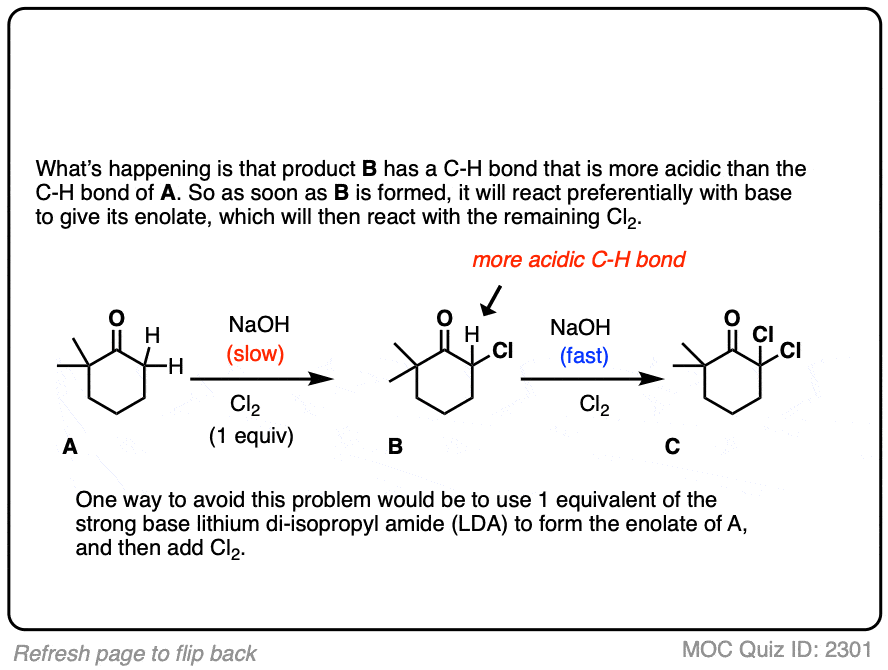
如果我们做一个烯醇化物在存在卤素如Br2,Cl2,或者我2,一个新的C-halogen债券将形成。这就是所谓的卤化烯醇化物。
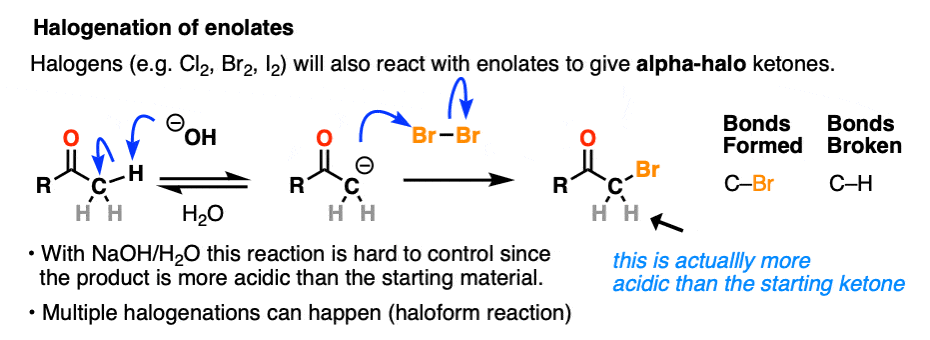
一个关键,要留意与卤化基本条件下,反应饼干怪兽特征(在吃一个饼干饼干怪兽永远不会停止)。
电子撤回组相邻碳氢键酸性更强,因为他们稳定负电荷。因此产生的产品将有更多酸性碳氢键,和更可能被基地。它会导致一个失控反应在多个卤素可以安装在相同的碳。(看到帖子:卤仿反应]
更好的(和更多的控制)方法是不可逆转地形成烯醇化物用超强碱乔治。然后添加(例如)Br2。没有失控反应。
醇醛反应
烯醇化物可以与其他醛或酮反应!这就是所谓的醇醛反应(看到帖子:醇醛反应)
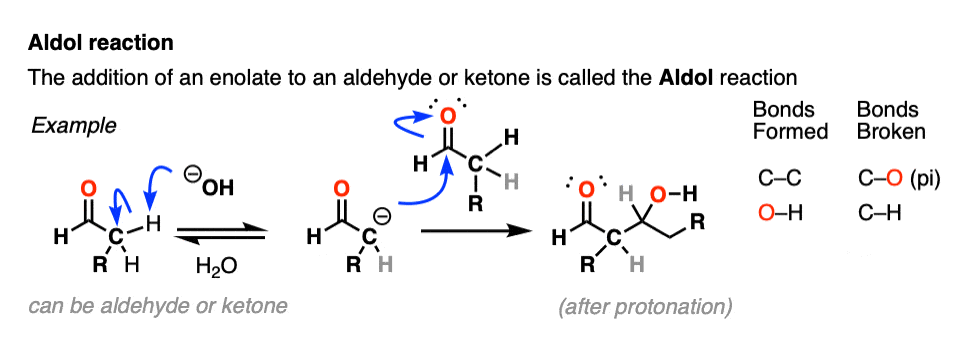
醇醛反应有许多表兄弟,由不同的名字像克莱森缩合,亨利反应,Knovenagel不管。他们都归结为同样重要的一件事:一个烯醇化物亲核试剂攻击一个羰基碳亲电试剂。
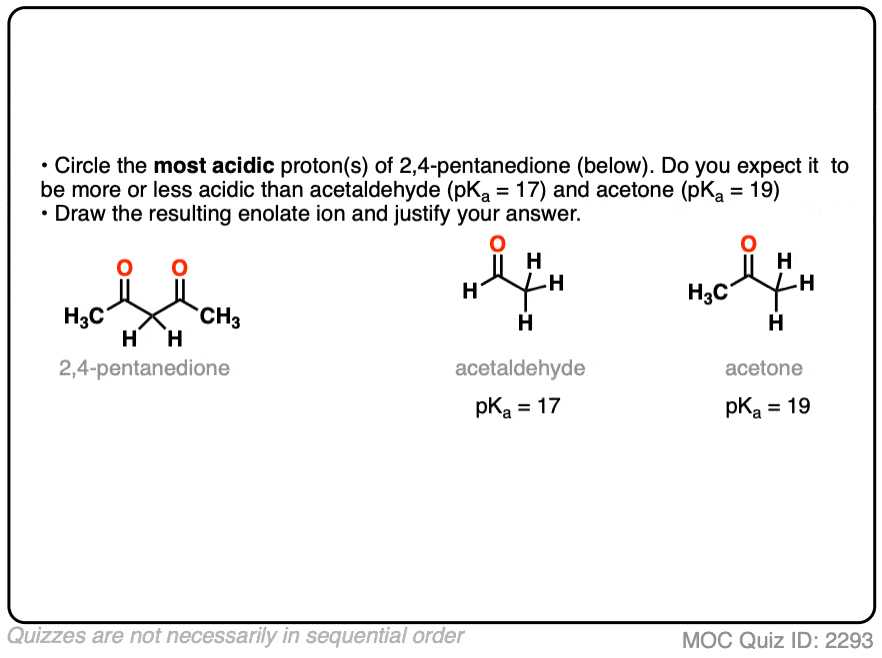
6。额外的吸电子集团使烯醇化物更稳定
所以如果醛和酮相当酸性(pK一个17日至19日)发生的酸度α碳当我们添加一个电子撤回组吗?
 点击翻转
点击翻转
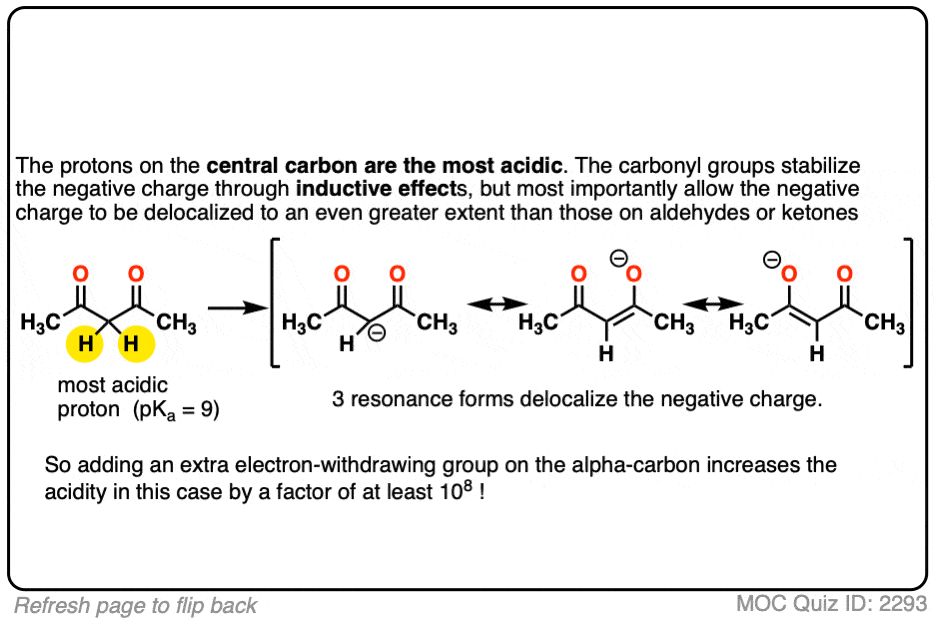
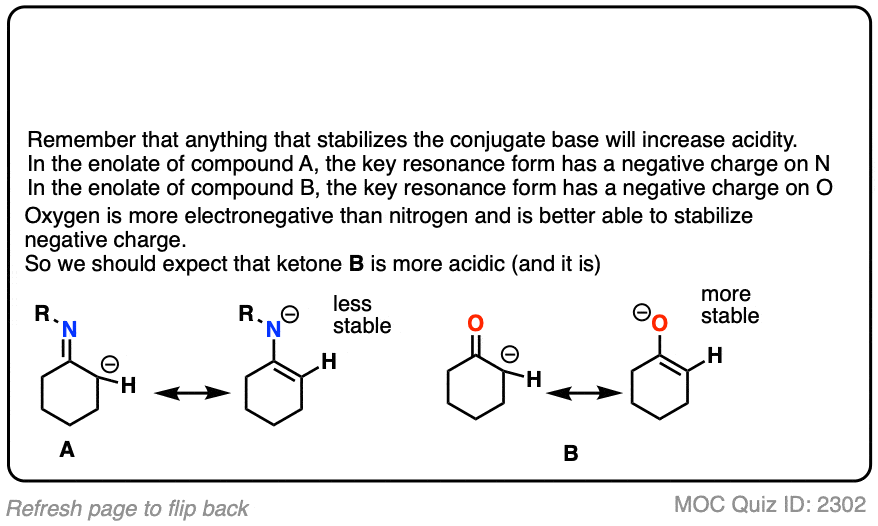
记住,任何因素,稳定共轭碱会增加酸度。所以它应该成为一个更强大的酸因为由此产生的负电荷共轭碱可以分散到多个氧原子也稳定了额外的诱导效应。
底线是烯醇化物,两个相邻电子撤回群体更容易形成!
例如,beta-keto酯类和乙酰乙酸酯类(pK一个大约11)可以很容易地deprotonated强碱如CH3CH2O (-) (注5]。
一个有用的反应的烯醇化物是他们将执行N2反应烷基卤化物,形成新的碳碳键。这叫做,“烷基化”。
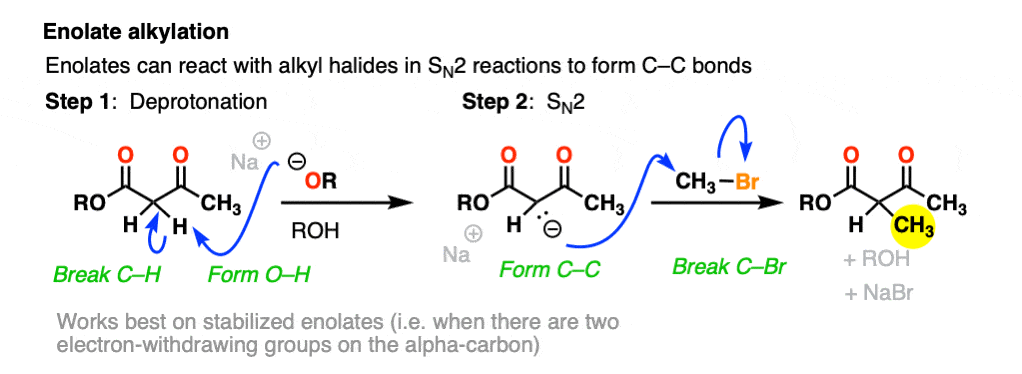
产品可以使失去有限公司2在一个过程称为(脱)丙二酸酯合成(di -酯类)或乙酰乙酸酯合成(beta-keto酯类)。(看到帖子:丙二酸酯和乙酰乙酸酯合成)
(注6- - - - - -关于这个反应与普通酮]
7所示。烯醇化物酯和酰胺
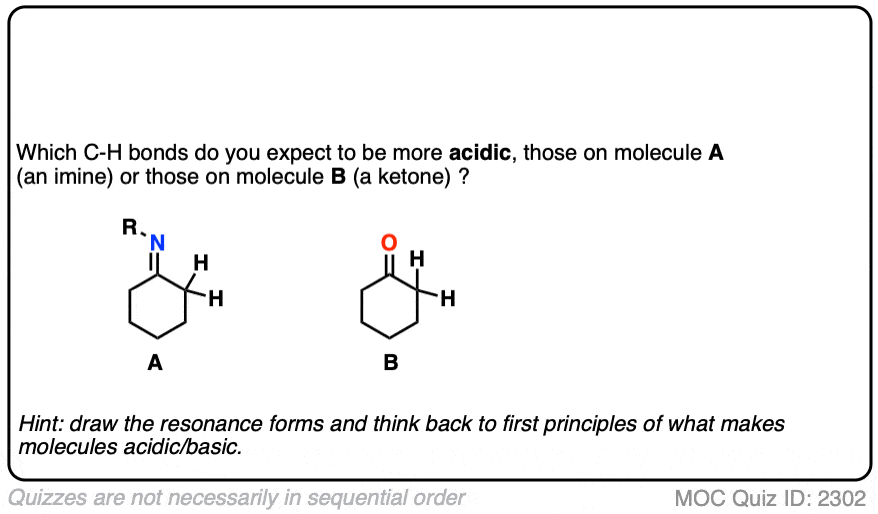
相反的方向,的酸度会发生什么α碳如果我们换一个酮对于一个酯吗?
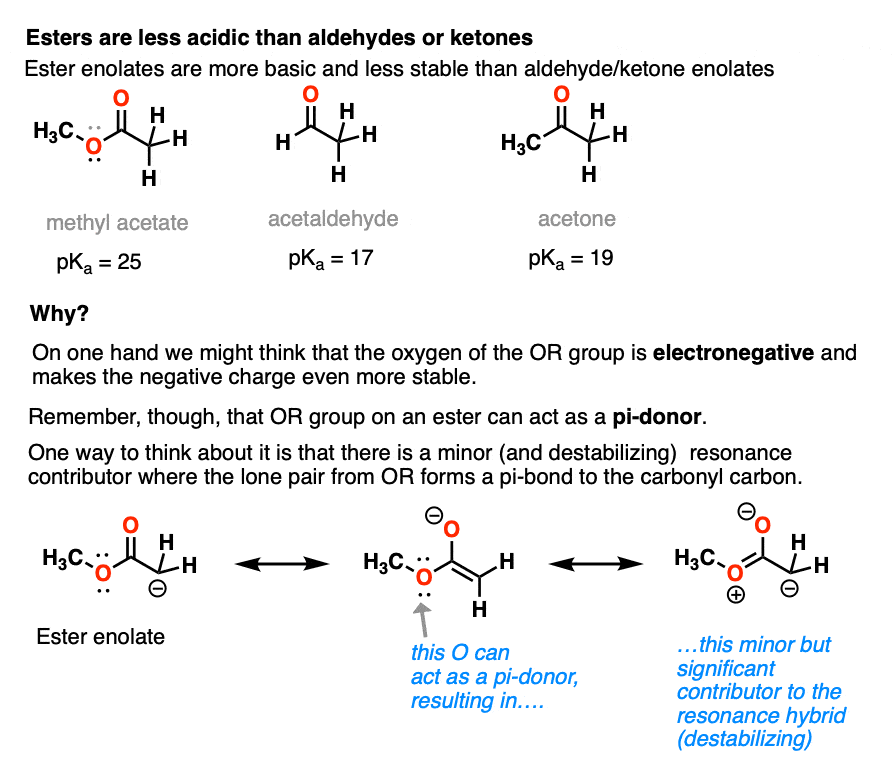
在的情况下酯的α碳实际上变成了减少酸性。因此酯烯醇化物是更不稳定醛和酮。
这或许令人惊讶。为什么酯烯醇化物少稳定吗?毕竟,我们不认为它应该是* *更好,因为它有吸电子或一组吗?
答案是哦,或者相关的原因激活组在芳环上而不是团体才会安静下来。
是的,氧的电负性,但孤意味着它也是一个pi-donor,能够形成一个切断的π键羰基碳。pi-donation超过电负性因素。
在某种意义上的C = O酯不能够稳定的负电荷吗烯醇化物因为烯醇化物阴离子是“竞争”的唯一的双或者组。
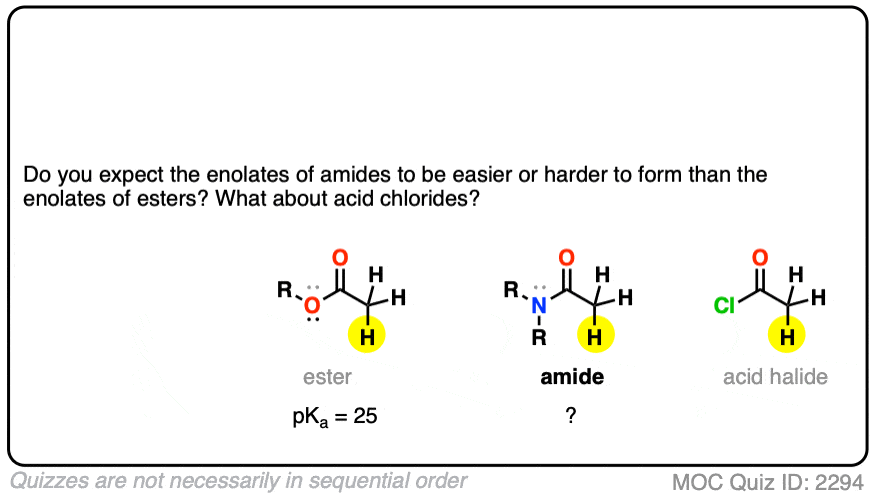
除了酯类,其他组织能够充当pi-acceptors如没有2、CN和酰胺也将形成烯醇化物。
知道你现在知道,你认为酰胺(NR的烯醇化物2)将更多的或少比的稳定酯(提示:回想激活组芳香环)
 点击翻转
点击翻转
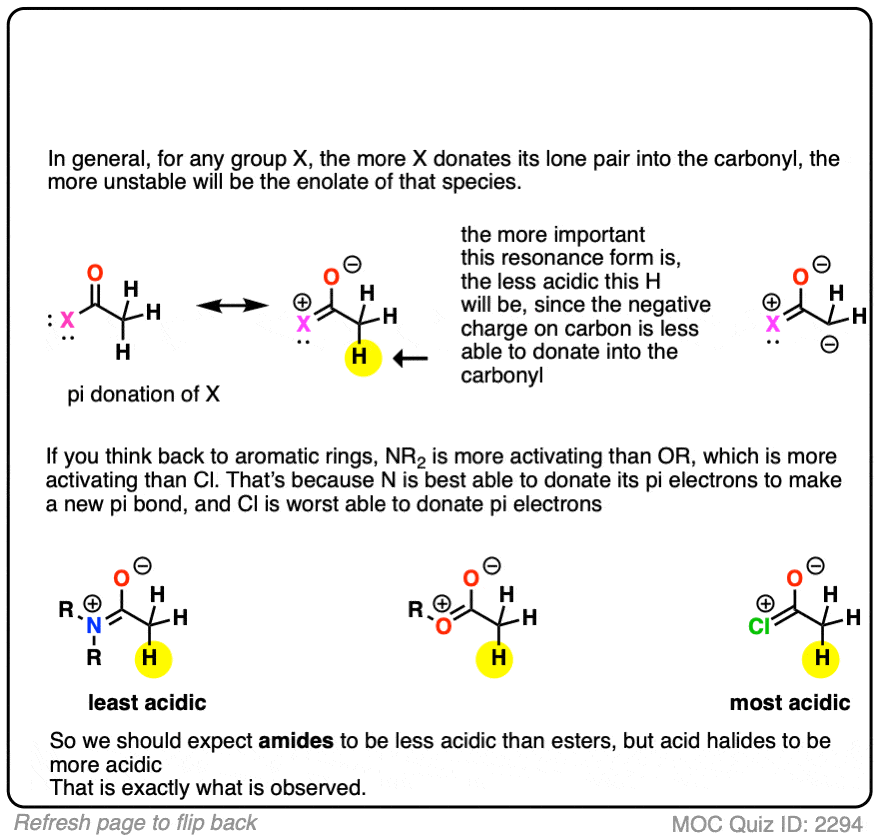
思考一个简单的方法是把C = O作为接受一个桶的孤对电子对碳。当已经有一个或一个NR电子供体2毗邻C = O,将会有更少的桶里的一对电子碳,和烯醇化物会更不稳定。
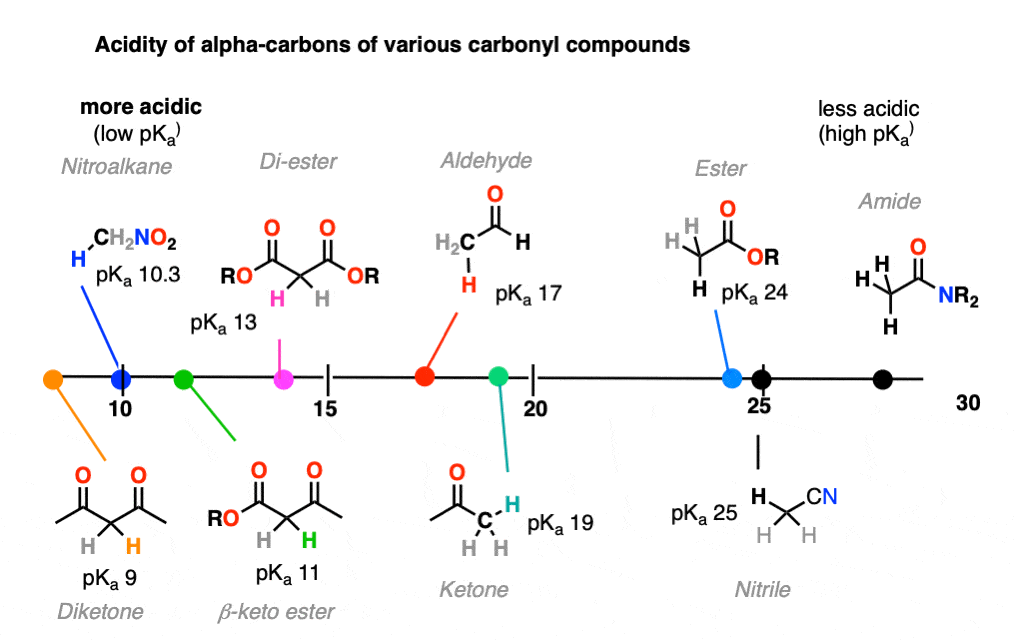
它可能有助于把这些趋势在这样一个图:
8。酮烯醇化物
本文不会不完整的简要提到的并发症酮烯醇化物。
在很多情况下治疗酮可能导致与基地两个不同的烯醇化物。
那么,我们如何知道哪一个“赢了?”。
我们把它以同样的方式与双键,即。扎伊采夫规则。就像烯烃,更多的代替烯醇化物将最稳定。
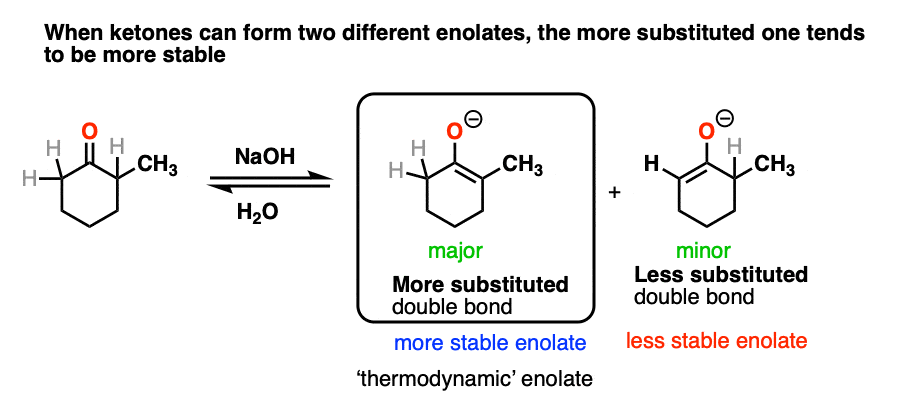
(请注意7]
因为这个原因代替烯醇化物被称为“热力学烯醇化物”——这是热动力更稳定。
然而,这种趋势形成了更多的替代烯醇化物会干扰我们的精心布置的计划!很多时候,我们想要形成一个烯醇化物在替换的越少酮“热”,不管它可能想要的。
解决这个问题的方法之一是趋势使用一个强大的,但是非常庞大的基地。基地di-isopropyl锂酰胺(乔治。)符合该法案。它就像一个大的锤头鲨,很难把鼻子伸进了严密的角落。所以,当看到一个酮绝大多数轴承两种不同α碳原子,它将移除一个附加到取代碳原子就越少。
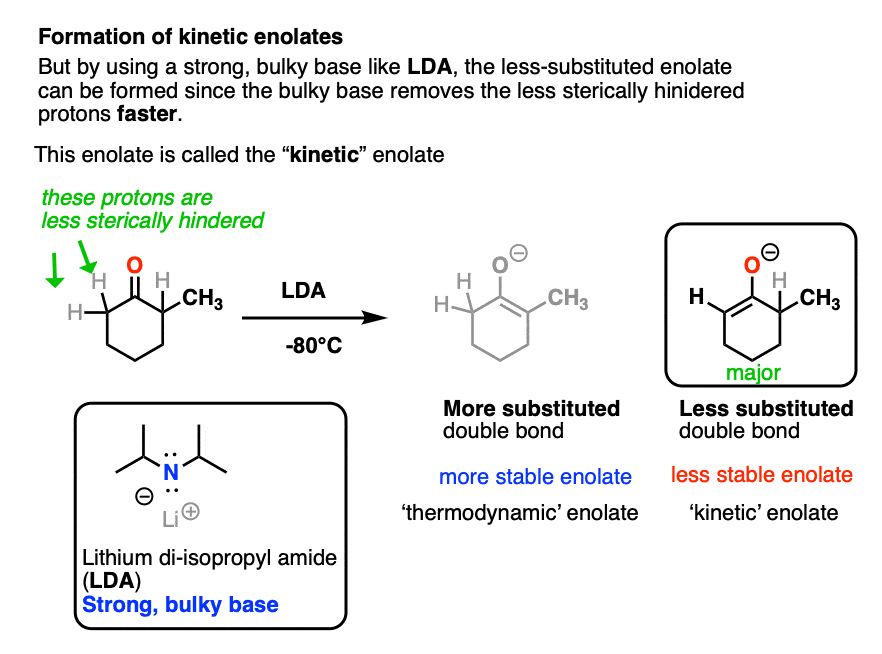
乔治。也用于制造的烯醇化物酯类、酰胺和腈,但是我们将会有更多的在将来的一篇文章中说。
9。总结
我们学到了什么?
- 烯醇化物结果从质子的迁移碳毗邻羰基(“α碳”)。他们由于归纳影响是稳定的,但更重要的是由于移位(通过共振)的孤对碳氧的电负性。
- 醛/酮没有一个碳氢键α碳被称为“非enolizable“因为他们缺乏酸性质子。
- 烯醇化物都是平。如果一个光学纯酮与手性α碳是治疗基地,当心失去光学纯度由于去质子化/质子化作用。
- Base-catalyzed酮烯醇醛和酮的互变现象经过一个烯醇化物。
- 烯醇化物将与卤素反应(例如Br2,Cl2,我2)醛/酮,烷基卤化物。
- 烯醇化物有两个电子撤回附加到组α碳特别容易形成吗。
- 烯醇化物的酯类和酰胺更加困难比醛和酮的烯醇化物形成。
- 在许多情况下,治疗酮与基地可能导致两种不同的烯醇化物。等强碱氢氧化钠或NaOR倾向于形成更多的代替烯醇化物因为烯烃稳定性随替换。
- 这种趋势可以克服使用强大的位阻基础像锂二异丙基酰胺(乔治。),形成了“动能”烯醇化物。
笔记
注1。我们可以计算出青睐这个反应是通过观察我们的酸碱反应的四个组件(酸、碱、共轭酸,共轭碱)比较pK一个酸的共轭酸。酸是醛(pK一个= 17)因为我们的酸是HO (-),共轭酸是H2O (pK一个= 15.7)。因此,平衡常数K(16 - 17 = 1)或10左右1产品的方向。所以很少烯醇化物将会出现在任何给定的时间。
注2。的醛碳氢键不是酸性,即使它似乎应该是!
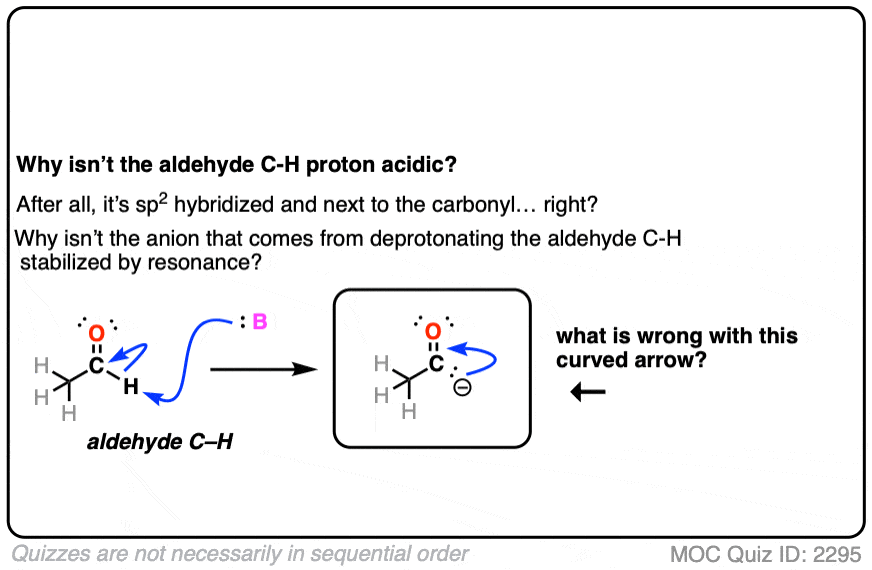 点击翻转
点击翻转
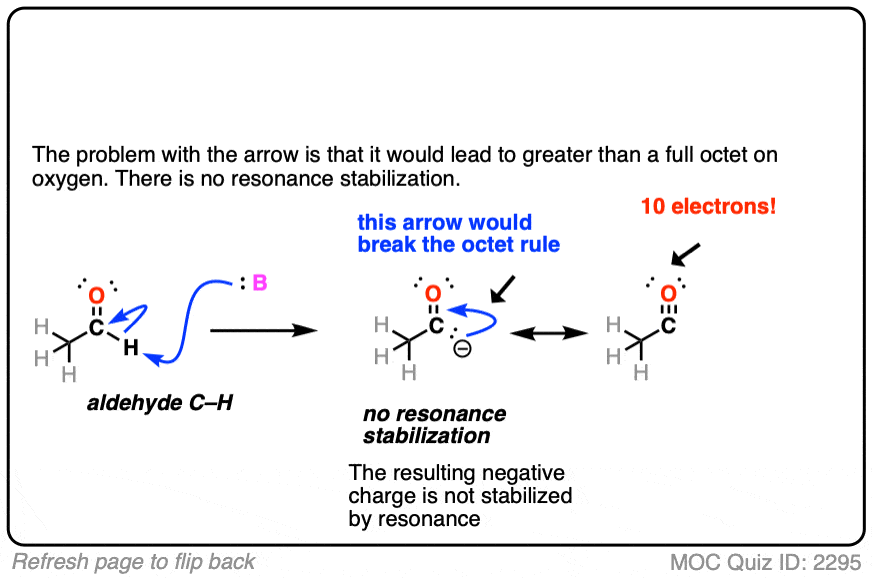
注3。在碳而不是氧烯醇化物往往反应。一个原因是,氧气会紧密地绑定到任何一个基地的抗衡离子使用(例如李+或Na +)。让氧气更被动的一个方法是使用碱金属盐大,形成一个较弱的离子键与氧气(如钾、K +)。
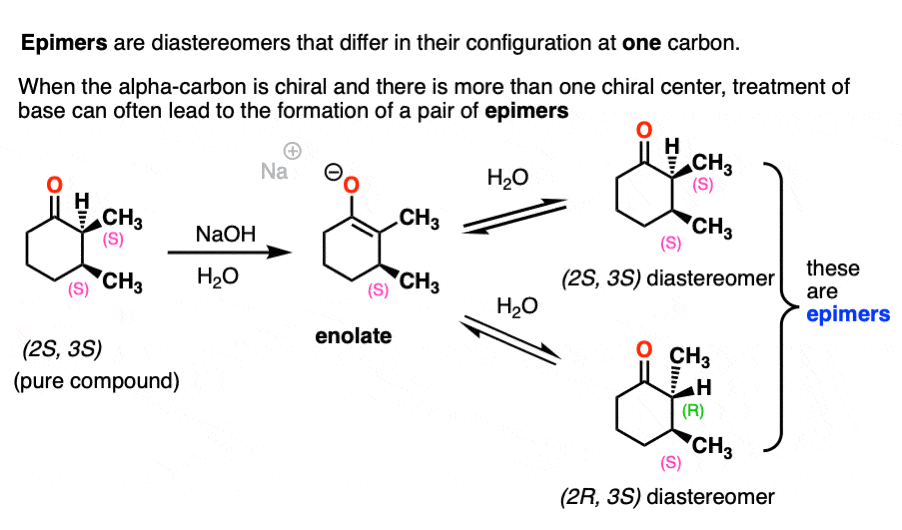
注4。当只有一个手性中心,它在α碳,结果将是外消旋化。当有多个手性中心,是之一α碳,只有上的手性中心α碳将受到影响。
这可能导致的形成异构体。差向异构体是我们给一个特殊的名字非对映体有不同的配置,只有一个手性中心。
例如,治疗这个光学纯(2 s, 3 s) -dimethylcyclohexanone基地将导致的混合物(2 s, 3 s) -dimethylcyclohexanone -dimethylcyclohexanone (2 r, 3 s)。他们不是对映体因为他们已经在颈- 3相同的配置。
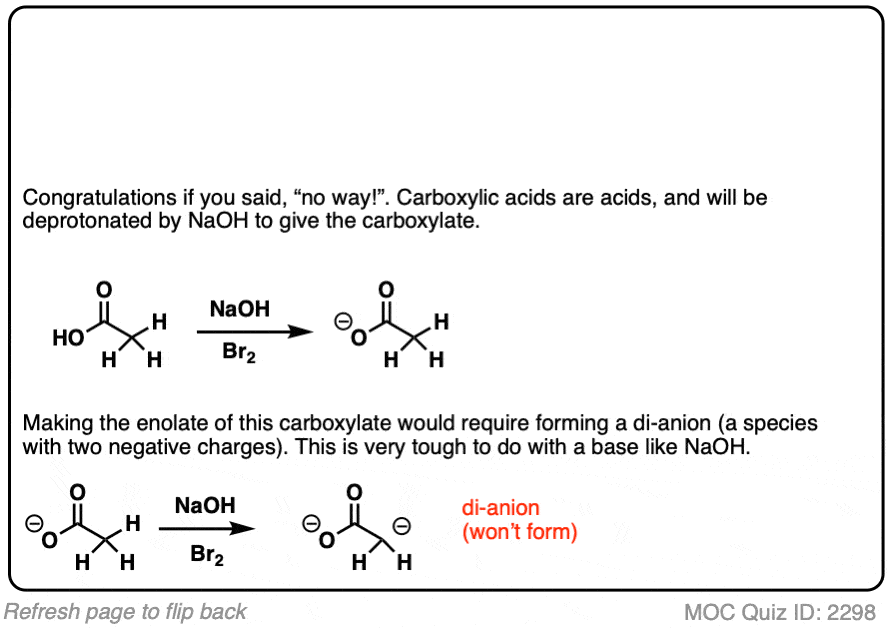
注5。我们必须小心我们使用的基础。如果我们用氢氧化钠,我们最终会执行皂化的酯(基本水解)。如果我们使用强碱与不同的R组我们可以最终执行酯交换。
注6。烷基化的普通使用NaOR基地和酮烷基卤化物等CH3我是一个大杂烩。它会导致多个烷基化发生。最好的办法是执行这些反应不可逆转地形成了烯醇化物使用强碱乔治。然后使用一个烷基卤化物。
这是经典的研究房子和Kramar。强烈推荐。
注7。典型的热力学的比例:动能烯醇化物在“热力学条件”(NaOR /卢武铉)约为4:1,但可以相差很大。第二页的这篇文章在形成热力学烯醇化物提供了一个很好的概述。
注8。我被问到的一个问题是为什么格氏试剂能够增加酮没有deprotonating第一个吃螃蟹的人。在所有的pK一个一个醛或酮大约是16和pK一个烷烃的大约50,所以为什么你不期望一个酸碱反应?
答案是,α质子是酸性的但他们仅仅是酸性的碳氢键与p轨道的切断π键。这是因为共振移位只能当p轨道可以重叠。
这使得去质子化碳相当慢,相对于除了到羰基也相对地的去质子化,不需要轨道重叠。
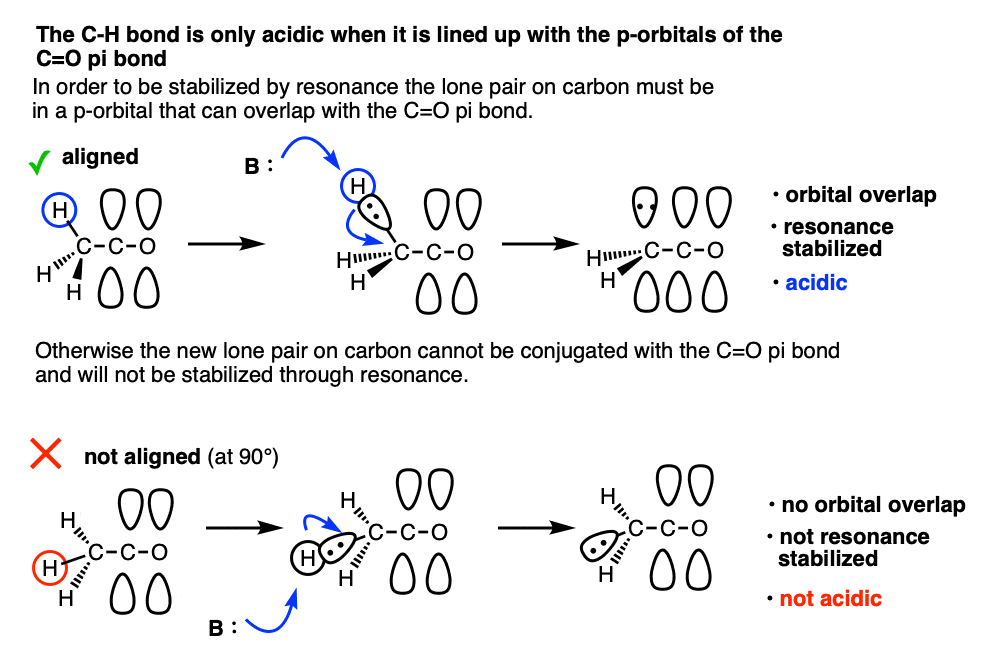
典型的例子是二环酮的桥头堡碳氢键。
注意9。两种其他的方法使我们遇到的烯醇化物介绍有机化学熊说。
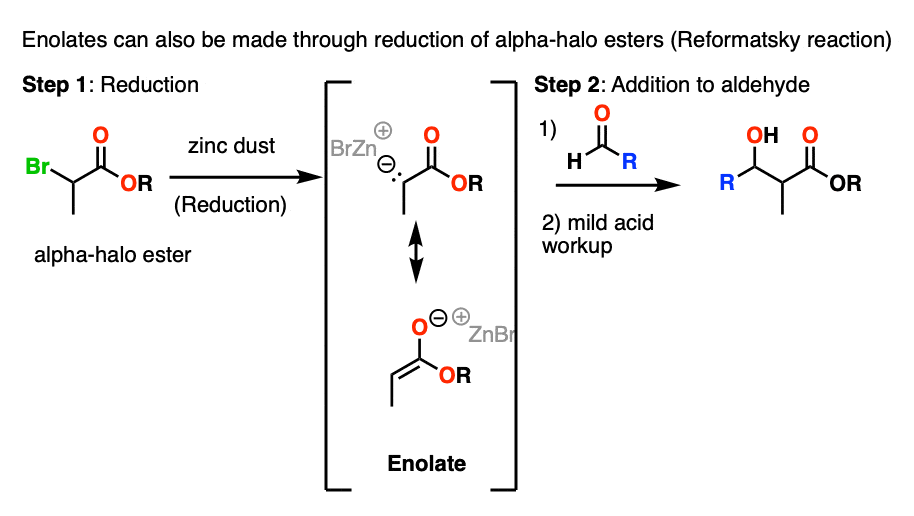
第一个是alpha-halo酯类可以用锌给烯醇化物这一过程被称为还原烯醇化物的形成。这是很像格氏试剂形成。
由此产生的烯醇化物可以添加醛和酮的反应称为Reformatsky反应。
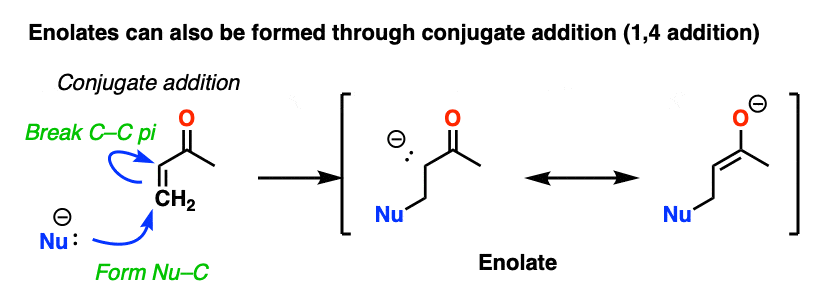
另一种方式的烯醇化物是通过共轭加成。添加亲核试剂到一个enone给一个新的烯醇化物。
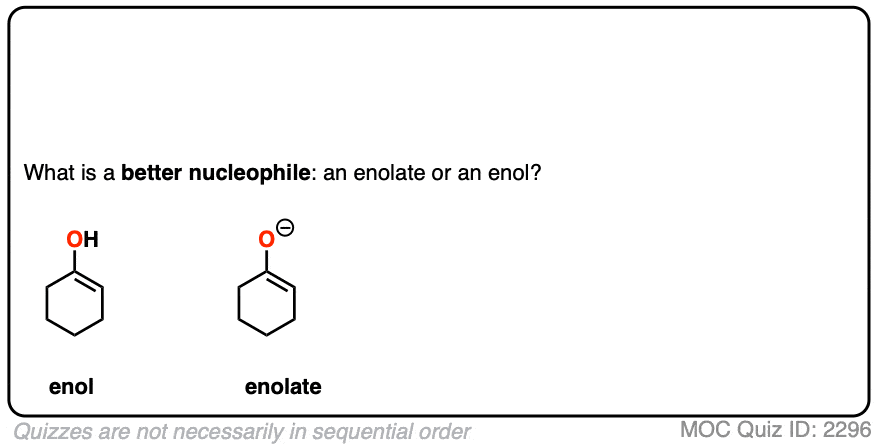
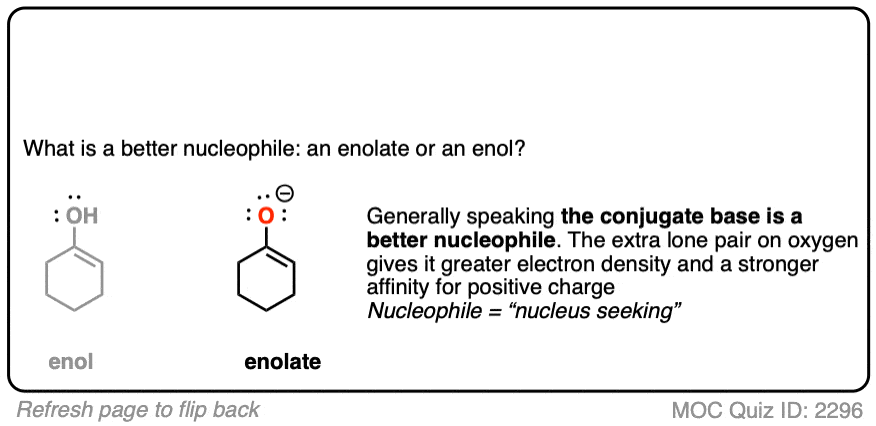
测试你自己!
 点击翻转
点击翻转
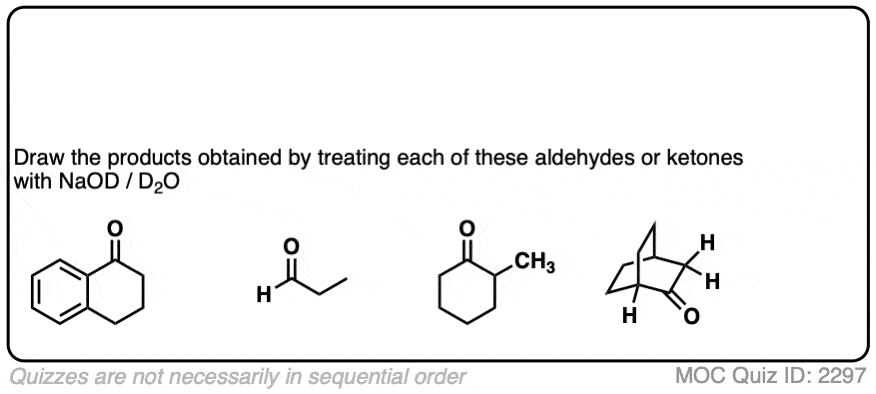 点击翻转
点击翻转
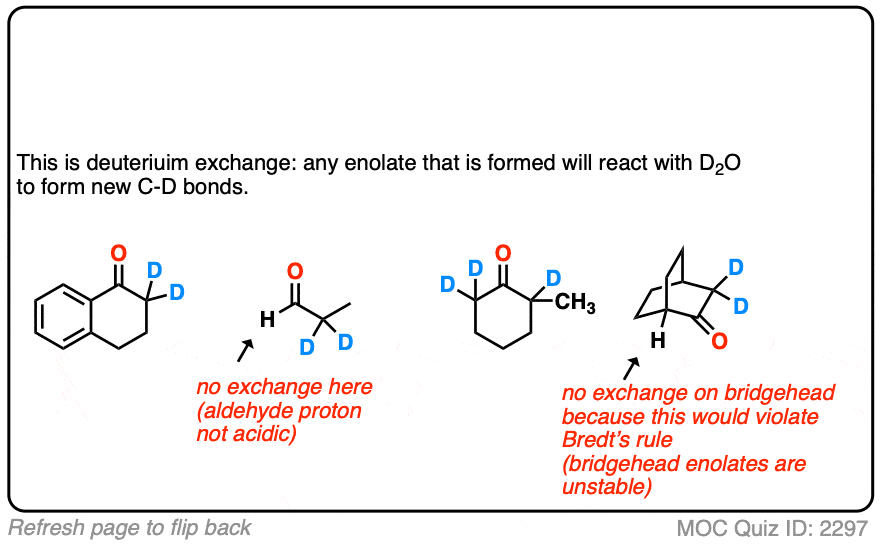
 点击翻转
点击翻转
 点击翻转
点击翻转
 点击翻转
点击翻转
(高级)引用和进一步阅读
引用
- 旋光性与互变异构的改变。第六部分。比较的外消旋化和溴化酮。
盛香港许和克里斯托弗·l·威尔逊
j .化学。Soc。1936年,623 - 625
DOI:doi.org/10.1039/JR9360000623
早期研究烯醇化物表明,外消旋化的速率等于卤化,暗示反应所得通过烯醇化物中间。 - 四面体报告25号:酮烯醇化物:regiospecific制备和合成的用途
吉恩·d天使
四面体1976年,32(24),2979 - 2990
DOI:10.1016 / 0040 - 4020 (76)80156 - 1
本文综述了各种方法烯醇化物形成,组成各种数据酮- - - - - -烯醇化物动力学和热力学conditions.Prof下形成的混合物。h . o .房子(麻省理工学院,然后佐治亚理工学院)发表了一系列论文和负碳离子烯醇化物化学,研究动力学和热力学烯醇化物详细的形成。这些论文的选择如下: - 负碳离子的化学反应。诉不对称酮的烯醇化物派生
赫伯特·o .房子和维拉Kramar
《有机化学》杂志上1963年,28(12),3362 - 3379
DOI:10.1021 / jo01047a022 - 负碳离子的化学反应。第九。钾离子和锂烯醇化物来自环酮
赫伯特·o .和Barry m . Trost房子
《有机化学》杂志上1965年30.(5),1341 - 1348
DOI:10.1021 / jo01016a001 - 负碳离子的化学反应。十五。立体化学4-tert-butylcyclohexanone的烷基化
赫伯特·o .房子,本·a·Tefertiller和休·d·奥姆斯戴德
《有机化学》杂志上1968年,33(3),935 - 942
DOI:10.1021 / jo01267a002 - 热力学和动力学控制的烯醇化物:一个项目面向问题的实验课
奥古斯汀对峙Jr .)迈克尔·a . Knopp和Jhong金姆
《化学教育1998年,75年(1),78
DOI:1021 / ed075p78
论文从j .化学。艾德。包括如何展示的概念动能和热力学烯醇化物在本科实验室会话。 - 负碳离子的化学反应。十六。立体化学的烷基化1-acetyl 4-tert-butylcyclohexane 1-cyano衍生品
赫伯特·o .房子和托马斯·m·光秃秃的
《有机化学》杂志上1968年,33(3),943 - 949
DOI:1021 / jo01267a003
在酮和酯烯醇化物,环外的构象上偏置环己烷环有一个小的偏爱亲电试剂的方法赤道方向。如果轴向进一步阻碍了取代基的增加,选择性增加。 - 一个立体选择合成(±)-gymnomitrol总额
史蒂文·c·韦尔奇,他Chayabunjonglerd
美国化学学会杂志》上1979年,101年(22),6768 - 6769
DOI:1021 / ja00516a057
这个总数的第一步合成的烷基化烯醇化物2-methylcyclohexanone,由去质子化乔治。在-78°C,紧随其后的是平衡的热力学更稳定的替代烯醇化物阴离子在室温下4 - 5小时。 - 具体的形成和烷基化烯醇化物阴离子的不对称酮:2 -苄2-methylcyclohexanone和2 -苄6-methylcyclohexanone
马丁胆和赫伯特·o .的房子
Org。Synth。1972年,5239岁的
DOI:10.15227 / orgsyn.052.0039
这个过程从有机合成有不对称的特定选择的烷基化的详细实验条件酮通过动能和热力学烯醇化物形成条件。 - 热力学和动力学控制的烯醇化物:一个项目面向问题的实验课
奥古斯汀对峙Jr .)迈克尔·a . Knopp和Jhong金姆
《化学教育1998年,75年(1),78
DOI:1021 / ed075p78
论文从j .化学。艾德。包括如何展示的概念动能和热力学烯醇化物在本科实验室会话。 - 动能烯醇化物形成锂Arylamide:碱度对选择性的影响
Keith Vanlandeghem Linfeng谢,库尔特·m·艾森伯格和伯尼尔卡洛琳
《有机化学》杂志上2003年,68年(2),641 - 643
DOI:10.1021 / jo0263465
本文的主题烯醇化物形成和检查的立体化学烯醇化物形成不同的性质取决于Li-amide基地。一些高级主题相关的论文烯醇化物化学: - 伪麻黄碱作为一个实际的合成手性辅助光学纯度高羧酸、醇、醛、酮
安德鲁·g·迈尔斯,科比h·杨,侯,莉迪亚,麦金斯(David j . Kopecky和詹姆斯·l·格里森
美国化学学会杂志》上1997年,119年(28),6496 - 6511
DOI:1021 / ja970402f
的烯醇化物来自N-acylation伪麻黄碱可以有选择地从脸部烷基化。它是容易诱发选择性很大在一个反应比构型,和一个办法是使用手性辅助——你一个手性的支架上,附着在基板上,然后删除在你烷基化。这是一篇论文,说明实际化学——当时,更容易获得比现在,伪麻黄碱和迈尔斯(现在哈佛大学)教授广泛记录的反应和程序,以使其可再生的,健壮的,可靠的。 - 不对称烷基化反应的手性酰亚胺烯醇化物。一种实用方法拆分.alpha.-substituted的综合羧酸衍生品
d·a·埃文斯·m·d·埃尼斯,d . j . Mathre
美国化学学会杂志》上1982年,104年(6),1737 - 1739
DOI:10.1021 / ja00370a050
这是一个里程碑式的论文,埃文斯教授率先使用手性助剂和展示了他们可以用于不对称醛醇反应。这里使用的辅助是一个手性oxazolidinone,这种化学被称为“埃文斯不对称醇醛”或“埃文斯手性辅助”。 - 无环stereoselection。7所示。立体选择合成2 -烷基3-hydroxy羰基化合物的醇醛缩合
克莱顿h .和查尔斯·t·Buse威廉·a·Kleschick约翰·e·孙和约翰·迈克尔·c·Pirrung兰佩
《有机化学》杂志上1980年45(6),1066 - 1081
DOI:10.1021 / jo01294a030
本文介绍了概念时醇醛反应的立体选择性烯醇化物添加到另一个羰基化合物。过渡态模型进行了讨论,在螯合离子的作用也说明。
评论
评论部分