家/为什么有机化学是困难的(2)
博客
为什么有机化学是困难的(2)
最后更新:2022年10月6日|
今天的文章是关于一个简单的反应可以导致各种并发症。
我说的是格氏反应在这里,尽管原则可以应用于其他的反应。
格氏反应很简单。它涉及一个格氏试剂——这是某种形式的碳,镁盐,增加羰基化合物如醛,酮,或酯。
酮和醛的反应很简单:格氏试剂的亲核试剂,他们增加了亲电羰基碳,打破了C = O键和形成酒精酸后补充道。
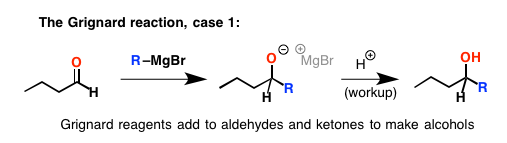
事情变得有点复杂酯类。如果您添加过多的格氏试剂,它增加了羰基碳。除了在这种情况下,它不会就此止步。之一新-氧原子上的孤可以重做一个π键与相邻的碳。这导致破损碳之间的债券,或导致其弹射和整体的形成酮。但这并不止步于此。现在我们有一个酮与格氏试剂,反应很好,我们添加第二个等价的,形成一个新的酒精(酸后检查)。
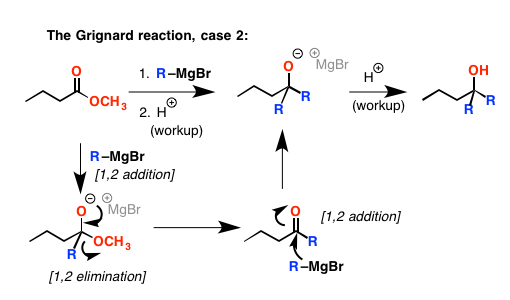
羧酸呢?你可能会认为他们的行为一样酯类。但是没有。格氏试剂是强大的基地,看到,当结合羧酸,他们是质子化了的。由此产生的带负电荷的羧酸盐(这是盐共轭碱的羧酸)然后几乎坚不可摧的亲核攻击由于强烈捐赠O(-)组。所以它就坐落在解决方案添加到酸,让我们回到我们的原始材料。

让我们回到酯类一分钟。你可能会认为如果你只添加一个相当于格氏试剂,你可以让它停下来酮阶段。嗯……不。发生了什么是速度消去反应速度非常快,比增加格氏更快酯——酮战胜的酯试剂。所以在一天结束的最后一个产品,两版的格氏添加到开始酯,一个相当于剩下的一半左右酯。
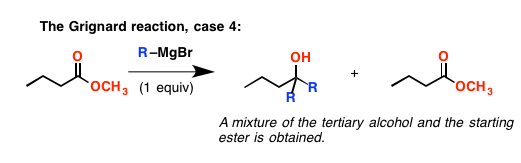
这就引出了下一个点:酮和醛反应比吗酯类。(反应性的顺序,顺便说一下,醛>酮> >酯]。所以如果你有一个分子酮和一个酯,你添加一个相当于格氏试剂,它将增加酮有选择地。
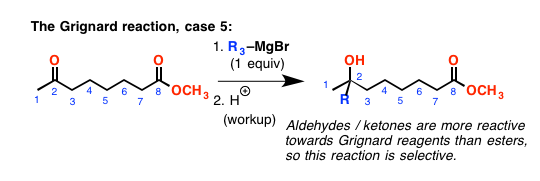
但小心!当你增加一个酮或醛,你成立一个新的醇盐(带负电荷的氧气)。这些都是良好的亲核试剂!如果醇盐氧是5或6键离开酯-如果它可以达到是可能的攻击酯羰基,做一个[1,2]添加/ [1,2]-消去反应形成一个内酯(循环酯)。
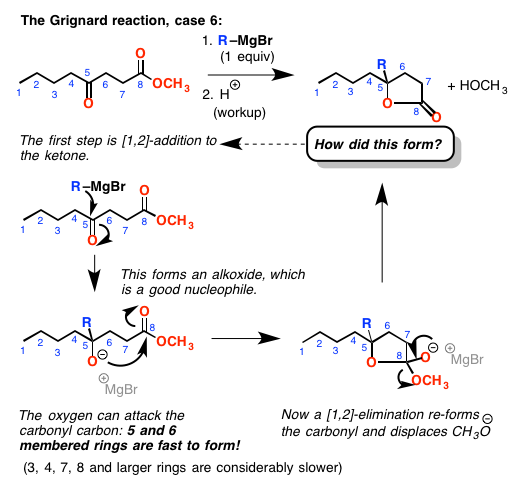
为什么5和6 -元环形成迅速,而3、4、7(和更高)员环不?很长的故事,但这只是它的方式。这是另一件你需要当心。
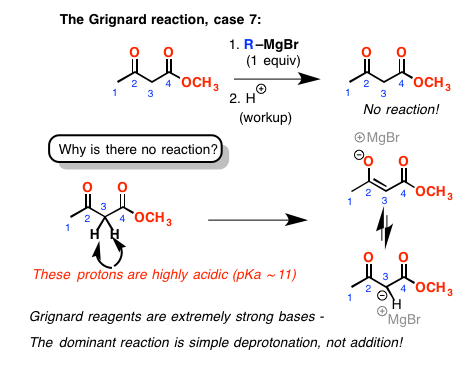
最后,我们来到这最后一个例子。我们有一个酮和一个酯。我们不能形成一个环。你可能会认为这只会增加酮在这里。再一次,不是这样。这一次,事实证明,在这两个羰基集团在近距离上的质子相邻CH2很酸。格林尼亚,一个强大的基础,简单地删除一个质子,不增加羰基碳。(像这样的另一个复杂因素:酮大多出现在他们烯醇形式,由于稳定氢键地和酯羰基氧气。]。酸性的检查之后,我们最终得到的原始材料。
教训:官能团和试剂可以在交互的方式,可以合理化回想起来,但该死的新手很难预测。这是什么使有机化学的一部分具有挑战性和令人沮丧,可以肯定的是,但也深刻而有益的。就是这样的人。
重点:
1)一些试剂的亲核试剂,行动基地。关键是当这是确定。
2)比酮醛更对的亲核试剂反应,活性比酯类。
3)格氏补充一个一次酯,不是不能休息的。
4)五和六元环的形成是快。
5)1,3-dicarbonyls异常酸性,这可以使格氏反应。
01焊接、结构和共鸣
- 我们怎么知道甲烷(CH4)是四面体吗?
- 杂化轨道和杂交
- bdapp.
- 轨道杂化和债券的优势
- σ键有六种:π键
- 一个关键技能:如何计算形式电荷
- 部分费用给线索电子流
- 四个分子间作用力以及它们是如何影响沸点
- bdappcom
- 如何使用电负性来确定电子密度(以及为什么不相信形式电荷)
- 介绍了共振
- 如何使用弯曲的箭头来交换共振形式
- bdapp1中国有限公司
- 如何找到最好的共振结构通过应用电负性
- 评估与负电荷共振结构
- 评估与正电荷共振结构
- 探索共振:Pi-Donation
- 探索共振:Pi-acceptors
- 总之:评估共振结构
- 画共振结构:3避免常见的错误
- 如何理解应用电负性和共振反应
- 债券杂交实践
- 结构和成键练习测验
- 共振结构的实践
02酸碱反应
03烷烃和命名法
04构象和环烷
05年有机反应的底漆
06自由基反应
07年立体化学和手性
08年置换反应
09年消除反应
11SN1 SN2 / E1、E2的决定
12烯烃的反应
- 烯烃E和Z符号(+顺/反式)
- 烯烃的稳定性
- 加成反应:消除的相反
- 选择性与特定的
- 在烯烃加成反应的区域选择性
- 烯烃加成反应的立体选择性:Syn vs反加法
- 马氏的HCl烯烃
- 烯烃Hydrohalogenation机制以及它如何解释马氏规则的像
- 箭头和烯烃加成反应
- 除了模式# 1:“碳正离子通路”
- 重组在烯烃加成反应
- 溴化烯烃的
- 烯烃的溴化:机制
- 烯烃加成模式# 2:“三元环”的途径
- 硼氢化反应,烯烃的氧化
- 硼氢化反应烯烃氧化机制
- 烯烃加成模式# 3:“协同”的途径
- Bromonium离子形成:一个(小)Arrow-Pushing困境
- 第四个烯烃加成模式——自由基
- 烯烃的反应:臭氧分解
- 简介:三个关键的家庭烯烃反应机制
- 钯碳催化加氢(Pd / C)
- OsO4(四氧化锇)Dihydroxylation烯烃
- m-CPBA (meta-chloroperoxybenzoic酸)
- (4)-烯烃合成反应地图,包括烷基卤化物的反应
- 烯烃反应实践问题
13炔的反应
14醇、环氧化合物和醚
- 醇-命名法和属性
- 醇可以作为酸或碱(以及为什么它重要)
- 醇的酸度和碱度
- 威廉姆森醚合成
- 威廉姆森醚合成:规划
- 从烯烃醚,叔卤代烃和Alkoxymercuration
- 醇通过酸催化醚
- 劈理的醚酸
- 环氧化合物醚家族的离群值
- 的环氧化合物与酸
- 环氧开环与基础
- 卤代烃与醇
- 甲苯磺酸盐和甲磺酸
- PBr3和SOCl2
- 消除反应的醇
- 消除醇与POCl3烯烃
- 酒精氧化:“强大”和“弱”氧化剂
- 阐明酒精氧化反应的机制
- 分子内反应的醇类和醚类
- 保护组醇
- 硫醇和硫醚
- 计算一个碳的氧化态
- 在有机化学氧化和还原
- 氧化梯子
- SOCl2机制醇烷基卤化物:SN2和SNi
- 酒精反应路线图(PDF)
- 酒精反应练习题
- 环氧化物反应测试
- 氧化和还原练习测验
15有机金属化合物
16光谱学
17二烯烃和MO理论
- 有机化学2中会发生什么
- 这些分子共轭吗?
- 结合有机化学共振
- π成键和反键轨道
- 分子轨道的烯丙基阳离子,烯丙基自由基和烯丙基阴离子
- 丁二烯的π分子轨道
- 二烯烃的反应:1、2和1、4
- 热力学和动力学产品
- 添加更多的1、2和1、4二烯烃
- s-cis和s-trans
- Diels-Alder反应
- 循环二烯烃和Diels-Alder亲二烯体反应
- Diels-Alder反应的立体化学
- 一昼夜的桤木挂式vs Endo产品:如何分辨它们
- 在一昼夜的HOMO和LUMO桤木的反应
- 为什么Endo vs挂式产品青睐Diels-Alder反应?
- Diels-Alder反应:动力学和热力学控制
- 复古Diels-Alder反应
- 分子内一昼夜的桤木的反应
- Regiochemistry Diels-Alder反应
- 应对和克莱森重组
- Electrocyclic反应
- Electrocyclic开环和闭包(2)——六个或八个π电子
- 一昼夜的桤木练习题
- 分子轨道理论实践
18芳香性
19芳香分子的反应
- 亲电芳香取代:介绍
- 激活和去活化组织在亲电芳香取代反应
- 亲电芳香取代的机制
- 昊图公司,Para -和元董事亲电芳香取代反应
- 理解邻、对位、和元董事
- 为什么卤素昊图公司-帕拉-董事?
- 双取代的苯:最强的捐赠者“赢”
- 亲电芳香取代(1)——卤化苯
- 亲电芳香取代(2)——硝化、磺化
- 东亚峰会(3)——傅克酰化和傅克烷基化反应
- 分子内的傅克反应
- 亲核性芳香取代(NAS)
- 亲核芳香替代(2)-苯炔机制
- 反应在“苄基的“碳:溴化和氧化
- Wolff-Kishner Clemmensen,羰基和其他减少
- 更多的反应芳香Sidechain:减少硝基和拜耳威利格
- 芳香族合成(1)——“操作”
- 合成的苯衍生物(2)-极性倒转
- 芳香族合成(3)-磺酰阻断组
- 桦树减少
- 苯的合成(7):反应地图和相关的芳香族化合物
- 芳香族合成反应和实践
- 亲电芳香取代实践问题
你的解释方式比我的有机讲师!感谢你让一切都那么简单!
好的,我很高兴你找到该网站帮助Nagina !
在上面的句子中开始“在一天结束的最后一个产品,两个相当于格氏添加了…”,这可能是更清晰的说“在一天结束的最后一个产品,两版的格氏添加到开始酯…”
这是一个很好的网站,保持良好的工作。
改变它。谢谢你!
你的网站非常清晰和有用! !我爱每一步显然是解释和颜色和数字是很容易遵循。谢谢你! ! ! ! !
谢谢!伟大的文章。感谢覆盖所有可能的细节。
如果α酮或醛氢pka约17日至19日,为什么不会负碳离子(非常强碱)文摘α氢。
一般来说,增加羰基是质子的速度比删除除非羰基位阻。不是快的一个原因是,碳氢键只有酸性的时候在一个键的构象与羰基的p轨道。
好文章!谢谢:-)
你好,如果一个格氏试剂在格氏反应形成二次酒精与二氧化碳发生反应,形成一个金属羧酸盐,为什么这边反应反应不会有问题吗?
从你这里我想我需要更多的上下文。你为什么要故意添加二氧化碳反应形成的二次酒精?
伟大的网站。抱歉教授的不工作,但(你知道)你的解释和详细的指出异常对我们极其有用的有机化学的学生。谢谢你!