消除反应
E1cB——消除单分子的共轭碱
最后更新:2022年12月2日|
- 的E1cB(单分子,消除共轭碱)机制是第三机械途径消除反应。
- 在许多方面,这是完全相反的E1机制,第一个一步是去质子化形成一个负碳离子,紧随其后消除在第二步。
- 它有时出现在介绍有机化学课程,特别是在机制醇醛缩合,芳炔形成,消除烯基卤化物给炔烃。
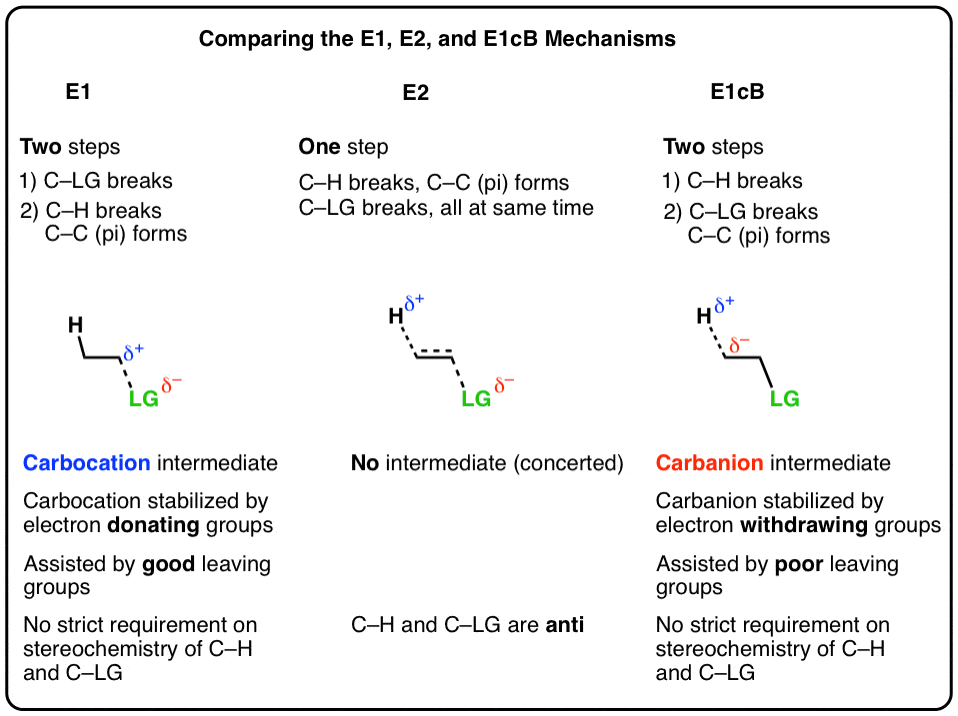
表的内容
- 消除给烯烃:E1和E2通路
- E1的cB机制
- E1cB机制和E1机制是两个极端,与E2机制躺在中间
- 介绍有机化学中的E1cB机制的例子:醇醛缩合
- 另一个例子:从烯基卤化物炔烃的形成
- 形成芳炔苯的卤化物
- E1cB反应的反应坐标图
- 摘要:当反应可能经历一个E1cb机制?
- 笔记
- (高级)引用和进一步阅读
1。消除给烯烃:E1和E2通路
消除反应是最常见的和强大的方法我们已经学会了烯烃(碳碳π键)烷基卤化物。
在消除反应提供了新的碳碳π键,bond-forming相同的一般模式和bond-breaking总是观察!
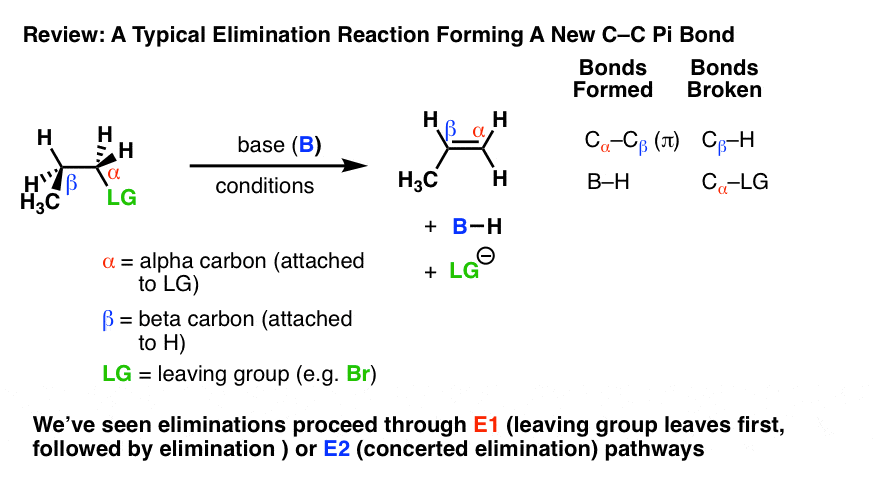
每个人都同意什么发生了。关键问题我们真的想知道,然而,是“如何它发生吗?”。
(编剧知道这一点。他们放弃的结局的电影——通常在第一个5分钟。但是是什么让你看正试图了解发生了一件事。为什么坐有人通过一个6小时的电影三部曲如果他们已经知道阿纳金最终将成为达斯·维达?)
这些债券的序列形式,打破“机制”的反应,或者如果你愿意,这个反应是如何发生的“故事”。
我们已经看到两个为消除这些模式到目前为止:
- 在E1机制,消除发生在两个步骤。C-LG债券最早出现,留下一个碳正离子。这是慢一步。在第二步中,β碳基deprotonates,打破碳氢键。碳氢键的孤对电子形成了新的π键的空的p轨道碳正离子。是单分子的(因此速率E1)因为它只取决于衬底。
- 在E2机制碳氢键和C-LG债券打破在同一时间形成新的碳碳π键。这是一个协同机制。双分子的(因此速率E2)由于反应速率都取决于基础和底物的浓度。
实际上是第三种可能,这就是我们今天要讲的。
2。E1的cB机制
第三可能性是,β碳deprotonated第一(b - h形式,打破碳氢键)。在限制的情况下,这将给一个阴离子中间,“共轭碱”的衬底。(参见:在有机化学酸碱反应]
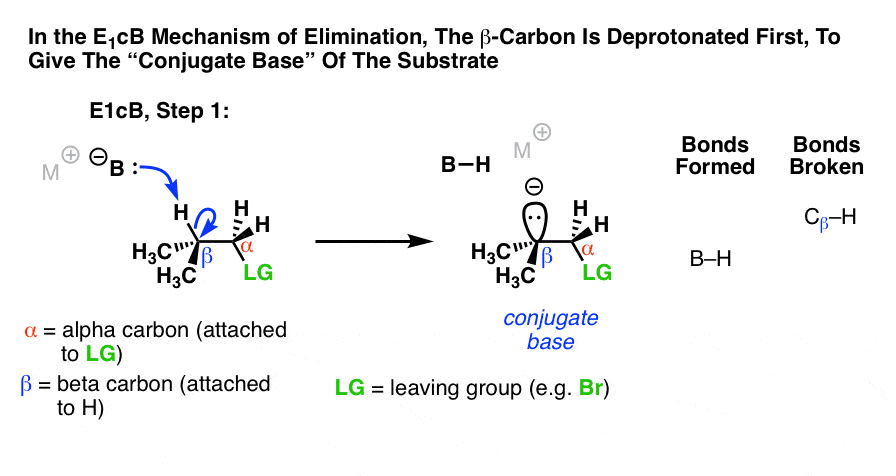
以来的第一步是去质子化给一个阴离子,你就会猜想是正确的这种机制是碳氢键时更容易发生相对的酸性,当α碳紧邻一个电子撤回集团像一个吗酮,腈(CN)或硝基(没有2)。这些团体稳定负电荷。(参见:稳定因素负电荷]
在第二步,的一对电子共轭碱然后替换离去基团,形成一个π键[形成碳碳π,打破C-LG]。
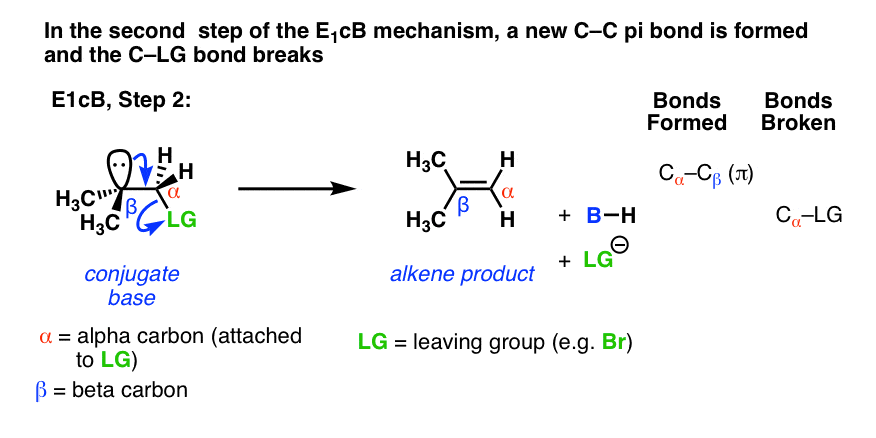
(许多)情况下的E1cB第二步是慢一步,你可以想象,这一步是由一个相对贫穷的慢离去基团喜欢(HO), (CH3O -)或者(信不信由你!)氟注1]。
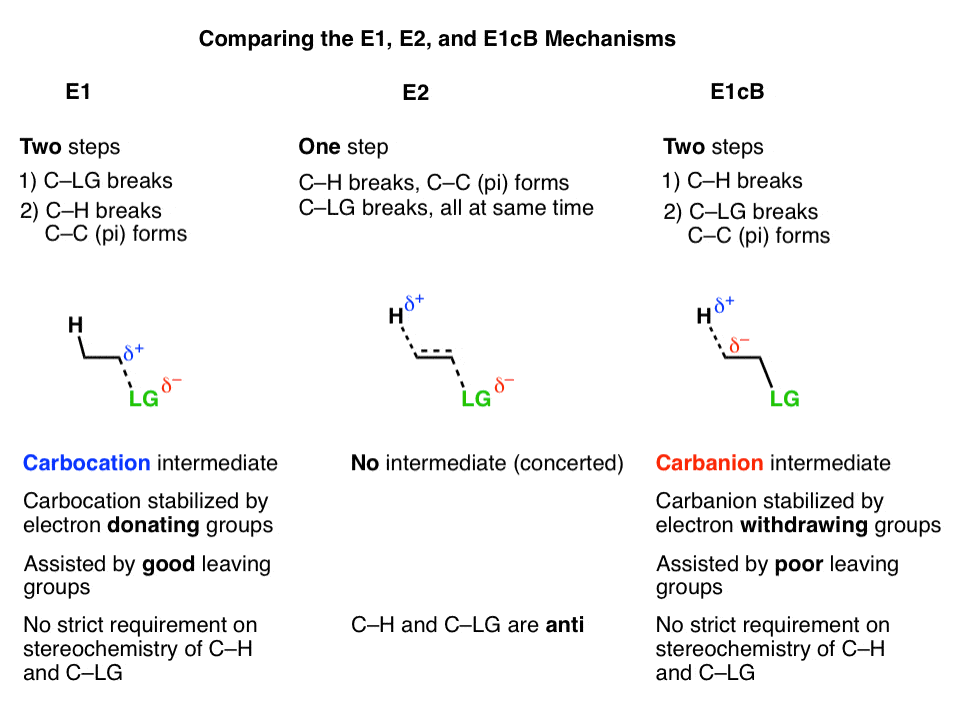
3所示。E1cB机制和E1机制是两个极端,与E2机制躺在中间
很有帮助的E1cB机制和E1机制是两个极端,或镜像,E2完全切割。这就像之间的连续“戒烟”和“被解雇”,与“相互同意分手”中间。
- 的第一步E1机制是一个损失离去基团(打破C-LG)给一个碳正离子中间,这一步是提拔当电子基组毗邻α碳(使碳正离子更稳定)。
- 的第一步E1cB机制是去质子化(打破碳氢键)给一个负碳离子中间,这一步是提拔当有吸电子集团相邻α碳(使负碳离子更稳定)。
- 的E2机制是在这两个极端之间,一切发生的地方。
那么这种机制实际上发生在介绍有机化学反应我们看到?是的它!
4所示。介绍有机化学中的E1cB机制的例子:醇醛缩合
最常见的例子E1cB在介绍有机化学机制醇醛缩合反应最后一步,特别是形成新的碳碳双键。(看到帖子:醇醛加成、缩合反应]
下面(左)是一个的产物酒精加成反应。如果这是与基础(如NaOCH加热3),它失去了水,给烯烃。(损失的水=“凝结”)
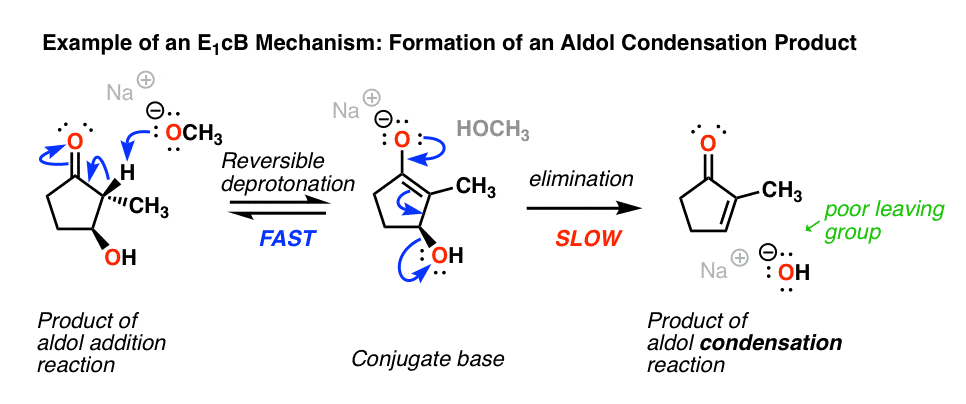
等一下,你可能会说。“我以为羟基离子(HO)是一个穷人离去基团吗?这是怎么回事?”
完全正确!如果有一个很好的离去基团(如OTs或Br)存在相反的哦,E2(甚至E1更有可能)机制。(注2]坏离去基团(HO)使消除步骤慢主要负责,这是一个两步机制。
另一个(非常微妙的)是立体化学。如果你看起来真的密切你会注意到在上面的图中,碳氢键和c哦债券并不反(要求E2),而是syn。
所以不像E2,E1cB机制没有严格要求碳氢键和C-LG债券是反的。
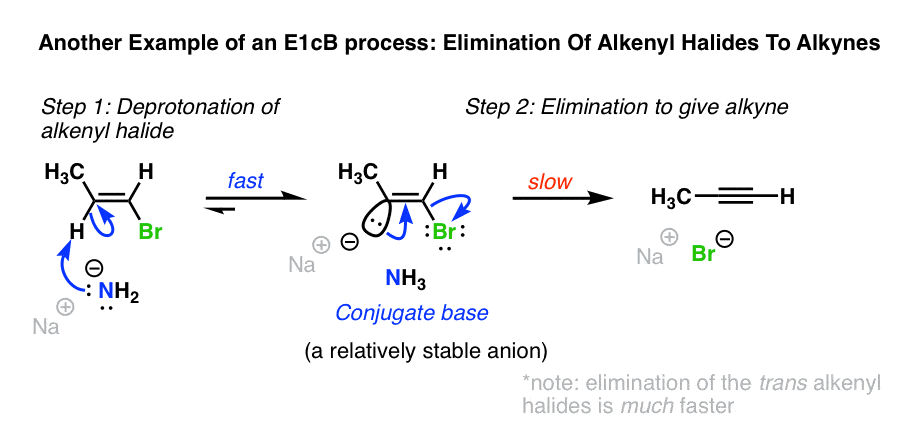
5。另一个例子:从烯基卤化物炔烃的形成
另一个例子的E1cB在的形成炔烃从烯基卤化物与NaNH2在液氨(NH3)。(参见:通过消除炔烃的反应]
当烯基卤化物与NaNH治疗2在北半球3作为溶剂,消除给炔烃发生。这个反应的作品即使H和Cl独联体彼此,似乎排除E2机制。(消除当H和卤化物反式要快得多,不过!(注3]
NaNH2是一个非常强大的基地。在sp2碳氢键的杂化烯基卤化物相当酸性(pK一个大约40)和能够稳定负电荷。
在这种情况下一个两步的机制可能是,1)负碳离子中间是由去质子化,其次是2)的损失离去基团。
6。形成芳炔苯的卤化物
一个相关的情况烯基卤化物的地方E1cB形成的机制会产生苯炔和其他“芳炔”[见:亲核性芳香取代苯炔机制]。尽管苯炔没有“真正”的π键,许多相同的原则的E1cB仍然适用。
去质子化的芳基强碱(NaNH氟化物2)首先,负碳离子。在这个特定的例子中,第二步是坏的损失离去基团,氟。(注1]
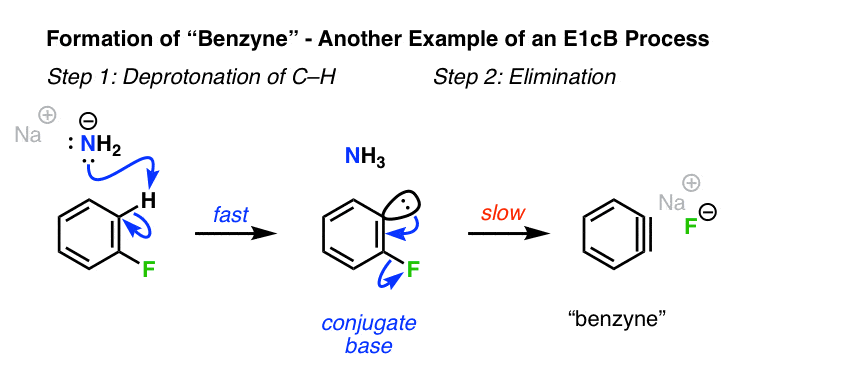
(请注意——显然,当F或Cl离去基团去质子化,是缓慢的一步。然而,当离去基团Br,或者我去质子化是cx快的速率决定和破损。请注意,这两个仍然符合E1cB只要通过负碳离子反应所得中间!]
7所示。E1cB反应的反应坐标图
那么的反应坐标图E1cB看起来像什么?如果我们假设第一步是快和第二步是缓慢(速度决定),有一个离子中间,的反应坐标图E1cB应该大致是这样的:
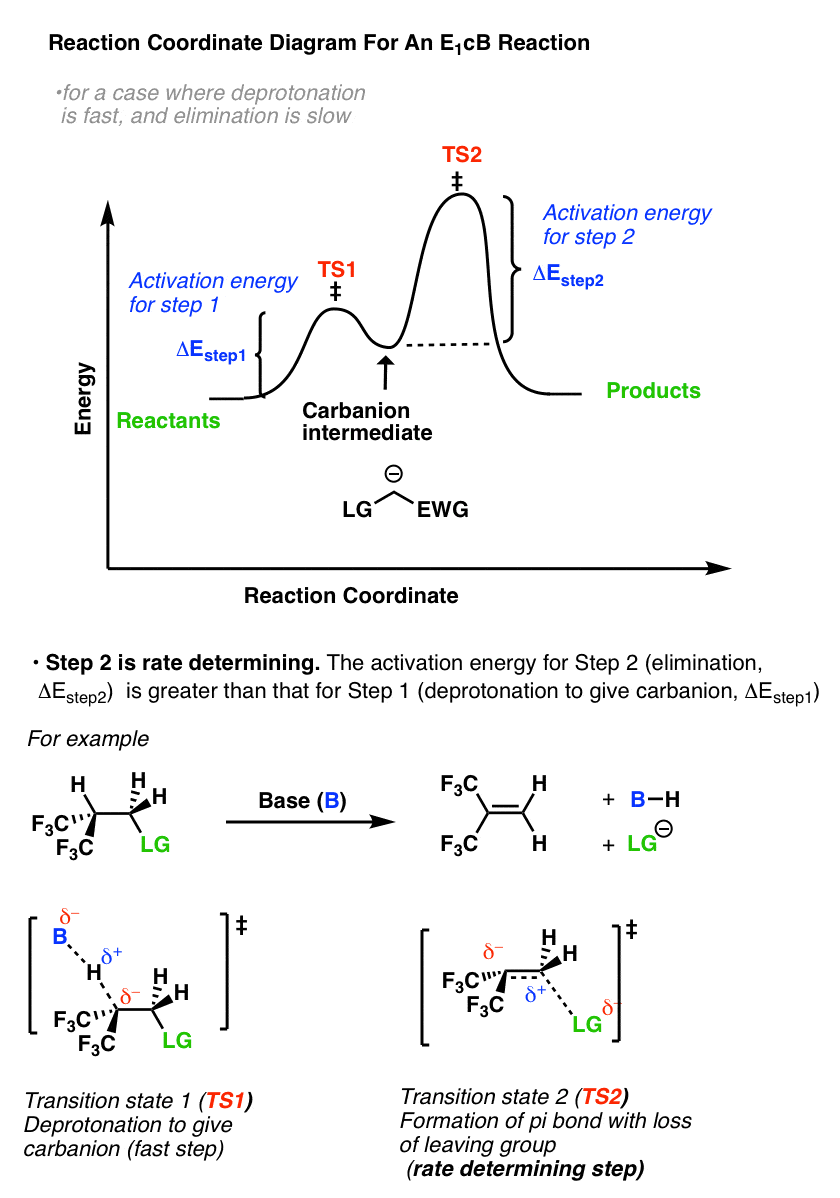
- 我们已经画了一个小峰从反应物到TS1与活化能(ΔEStep1)=[ΔE壹空间-ΔE反应物]。
- 然后继续阴离子中间(中间的“好”)
- 进而通过TS收益2与活化能(ΔE步骤2)=[ΔETS2-ΔE中间]
- 自从ΔE一步2>ΔEStep1,第二步是利率决定
比较的反应坐标E1消除,这是镜像。
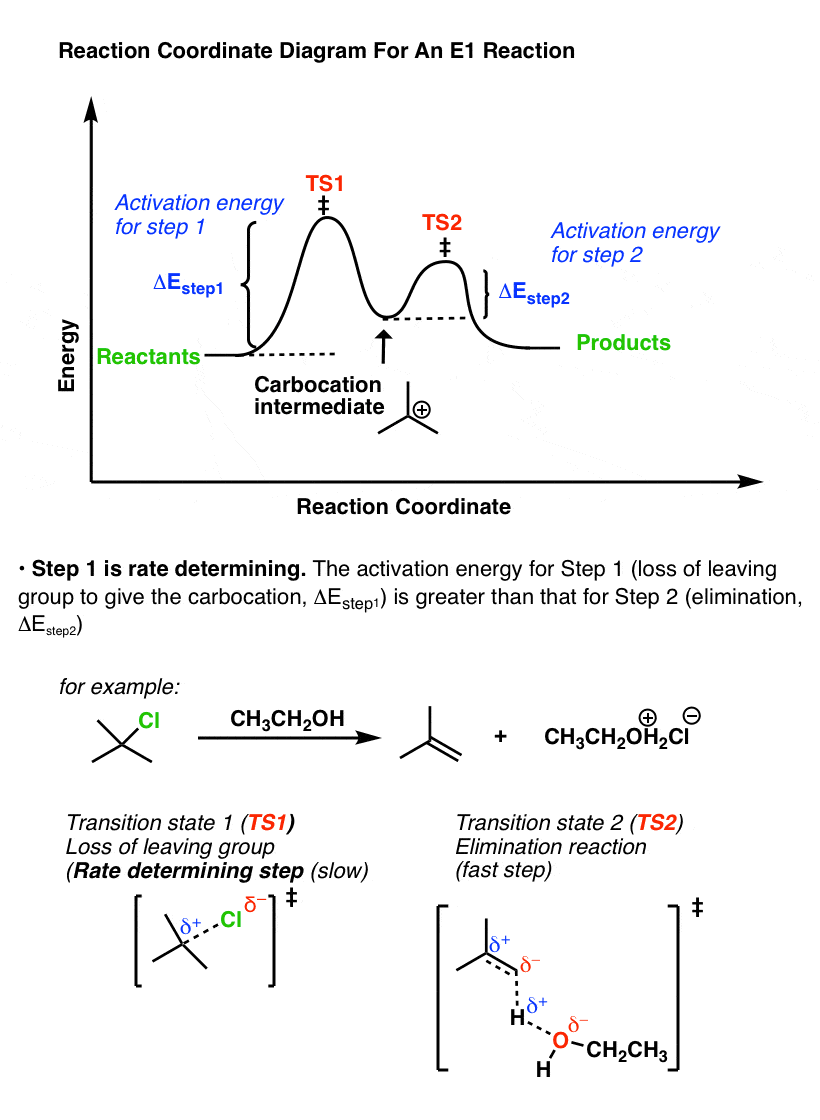
在E1机制、能量势垒(活化能,ΔEStep1(损失)的第一步离去基团)高于能量势垒(ΔE步骤2),第二步(消除)。
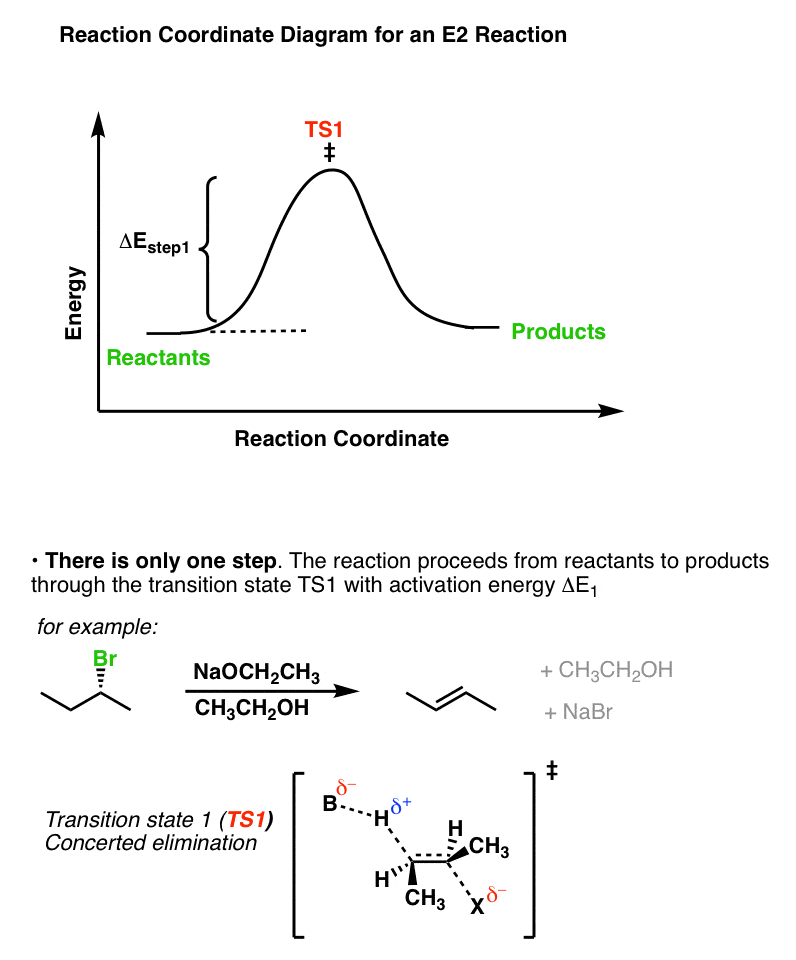
的反应坐标E2案例展示了这两个步骤同时发生。
8。摘要:当反应可能经历一个E1cB机制?
所以,总而言之,在E1cB机制,消除发生在两个步骤:
的E1cB机制在介绍有机化学不是很常见,但当它是观察,期待看到其中的几个特点:
- 一个吸电子集团的存在有助于稳定一个中间阴离子
- 一个相对贫穷的离去基团(这将减缓第二步)
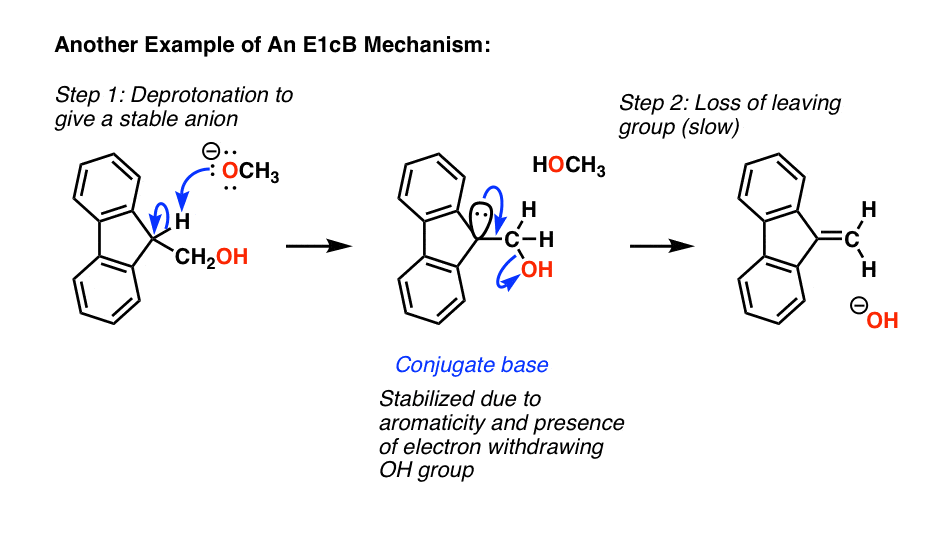
这是最后一个经典例子一个E1cB去质子化,导致阴离子(这是一个非常稳定芳香),慢慢地失去了穷人离去基团(氢氧离子)形成新的碳碳π键。
笔记
注1。我们从来没有考虑F (-)离去基团的年代N2的反应,但在某些情况下,它可以是一个离去基团在各种类型的替代和消除反应。一个例子是在亲核性芳香取代机制的另一个例子,通过一个相对稳定的离子。
注2。在这看到更多参考7,这表明在取消好了离去基团,E2之后,即使是一个强大的电子退出群。
注3。看到参考5——当H和Br的反应是容易得多反式彼此(可以用氢氧化钠!),但需要NaNH2 / NH3当Br和H独联体。
更多的E1cB
(1——我们决定E1cBvsE2- - - - - -2动力学同位素效应,3E1CB可逆的- - - - - -4E1CBIRR- - - - - -5E1CB阴离子- - - - - -6总结,总结了三个主要口味的E1CB- - - - - -7——立体化学。]
1。我们如何确定一些反应遵循一个E1cB机制与E2机制?你怎么知道区别呢?
这是一个很好的问题。
这些研究通常进行实验观察,其次是应用的好奇心。例如,考虑下面的反应。见参考9我们可能会天真地开始考虑这一个E2的反应。毕竟,有一个强大的基础,离去基团(坏虽然可能)和立体化学的碳氢键和c哦债券能够反。
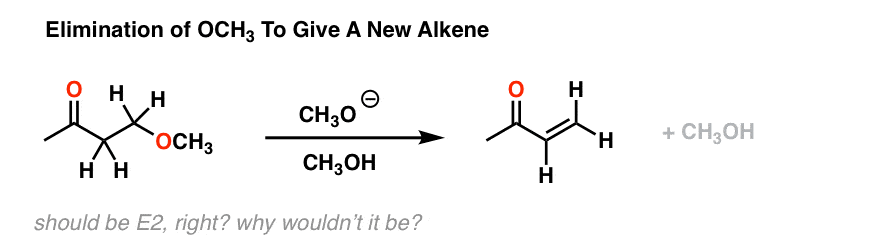
E2,对吧?然而,事实证明,如果你在氘运行这个反应溶剂,中途停止它,你最终得到的很多:
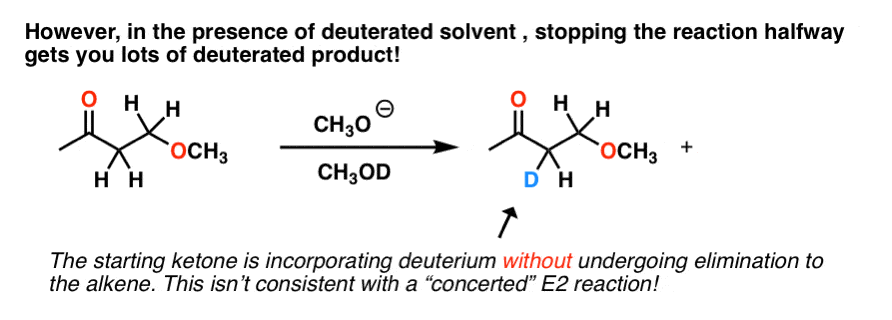
我们没有看到这个E2反应,因为碳氢键只能休息的时候反到离去基团。
在这个例子中,碳氢键显然是打破,但消除并不总是发生。那么到底是怎么回事?
最好的解释发生了什么是,基本形成了共轭碱基质,这可能与氘反应溶剂(如酸),导致氘交换在那个位置。
事实证明,交换比消除快220倍。
这是非常有趣的。但是,是多么重要共轭碱在消除?仅仅因为我们知道它的形成并不证明参与消除。
也许形成阴离子的只是一个“死胡同”,和一个慢E2负责的形成反应烯烃吗?
也许。它实际上需要相当多的努力回答这个问题(看到参考2),但是明确的证据表明,一个阴离子形成至少应该怀疑的种子E2是这里的唯一的可能。
2。动力学同位素效应
理论上,一个方法告诉之间的差额E1cB和E2是通过研究速率定律。事实证明,这通常是困难的E1cB(看到参考1),所以化学家们不得不采取其他方法。
的一个关键战略研究有机反应机制来衡量动力学同位素效应,利用同位素之间的细微差别在债券的优点。例如c - d (氘)债券略短(强)比碳氢键。如果碳氢键断裂发生的速率决定步骤,然后碳氢键化合物的反应速率(kH)应该略高于的速率氘将模拟用c - d键(kD)。这种差异,称为主要动力学同位素效应已被实验确定范围kH/kD在大多数情况下,2 - 8E2。
所以如果速率决定步骤碳氢键坏了,然后我们应该看到一个主要动力学同位素效应2 - 8的范围。如果它不是,那么这两个应该没有区别。
对吧?
3所示。E1cB可逆的
在上述反应中,动力学同位素效应约为1.0。这是清楚地表明,碳氢键不是破碎的速率决定步骤,这就排除了E2(共同)机制。
这些类型的E1cB反应被称为E1cB可逆的指示一个可逆去质子化步骤之后(慢)的损失离去基团。
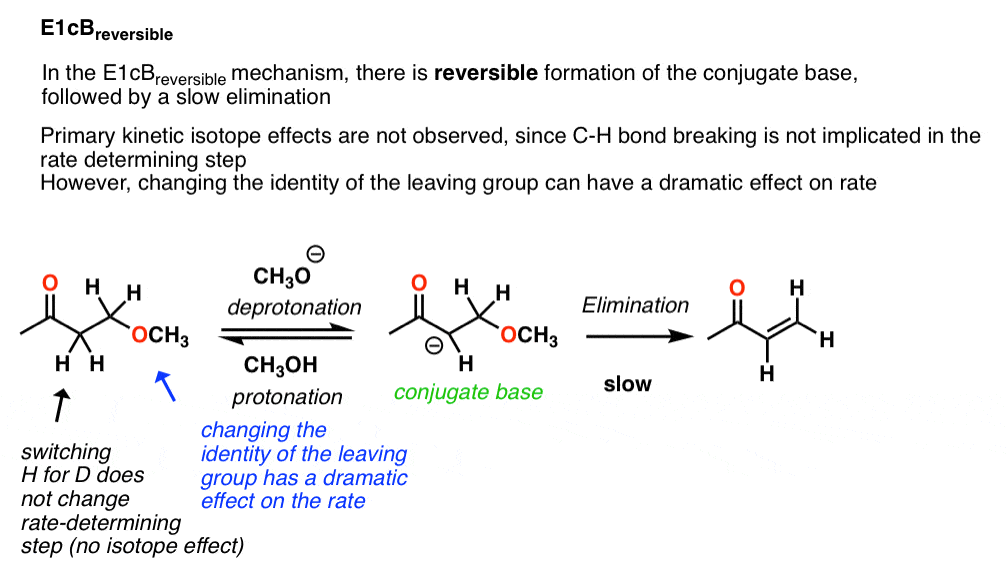
因为损失离去基团是缓慢的,应该期待看到一大对反应速率的影响通过调整的身份离去基团。例如,离去基团能力可以大大修改组X的身份苯基组(如图所示)。改变X集团强大的电子集团像NO2应该撤军离去基团更少的基本(和一个更好的离去基团),从而增加率。这就是发生了。
4所示。E1cB IRR
是它?只是衡量动力学同位素效应,我们可以区分E2和E1cB吗?
不一定!
例如,下面这个反应显示了一个大的主要动力学同位素效应,也就是说,碳氢键坏了的速率决定步骤。
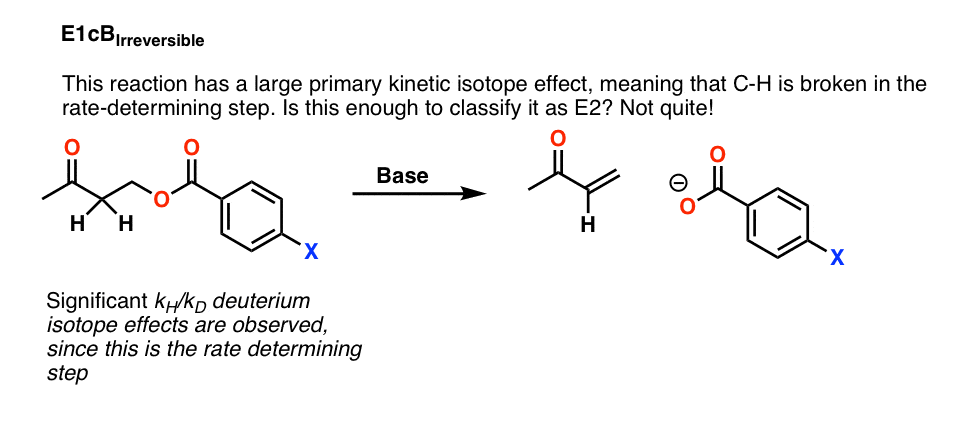
这是否意味着我们可以安全地分类这个反应E2吗?还没有。
这是可能的,这是一个一个E1cB去质子化的反应共轭碱是慢一步,和损失的离去基团是快。这就是所谓的E1cB不可逆转的
如果是这样的话,这是一个相当简单的方法来测试:改变离去基团。
打破C-LG债券的速率决定步骤的一部分E2,但不会的速率决定步骤E1cB。
事实证明,改变离去基团没有导致任何重大改变反应速率。
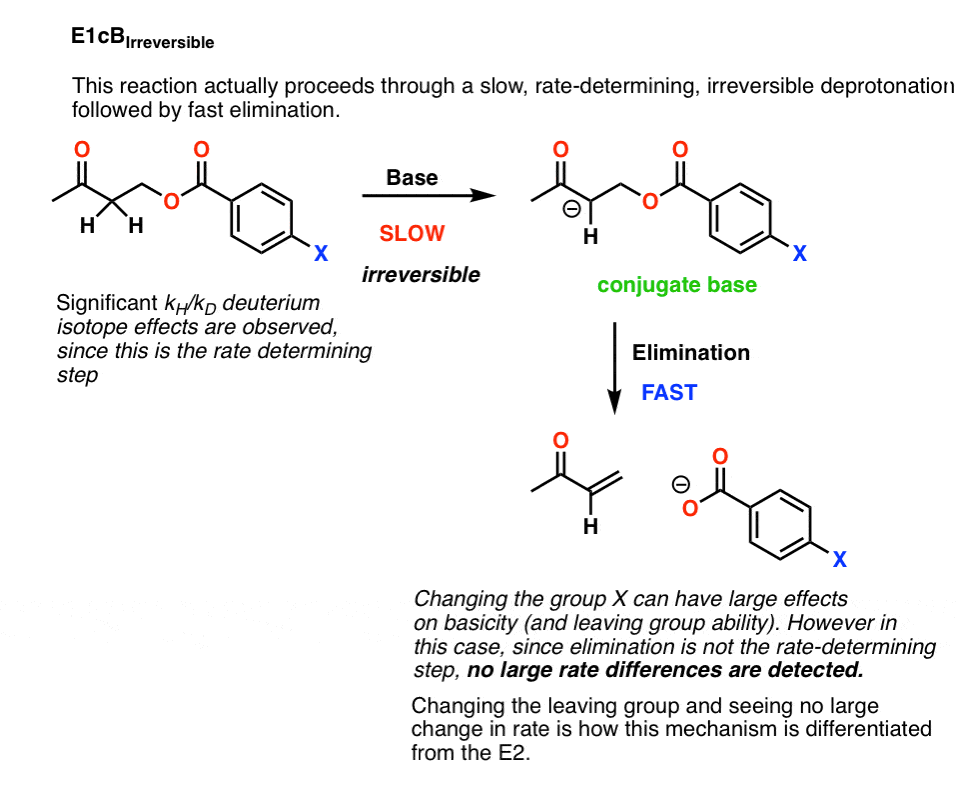
这将排除E2。
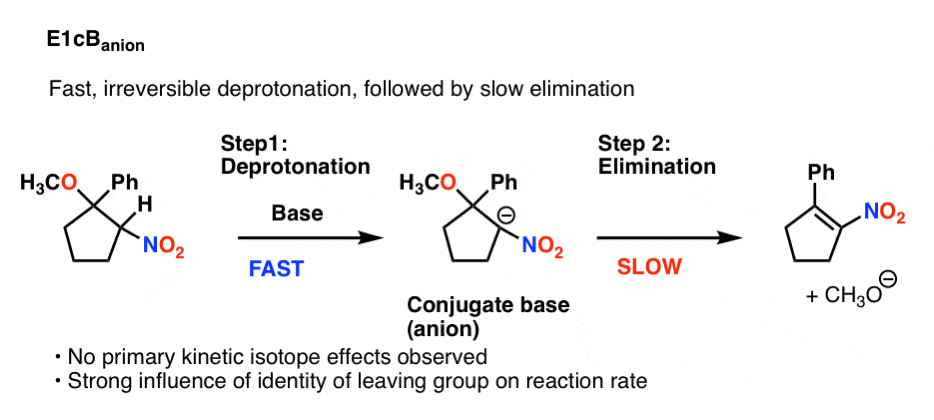
5。E1cbAnion
有一个主要的可能性了,这可能是最简单的可视化:快速、不可逆转的去质子化,其次是缓慢的消除。
这也不显示主要同位素效应,因为碳氢键断裂不是速率决定步骤。反应速率的身份很敏感离去基团然而,。
这味道E1cB被称为E1cB离子。下面是一个示例(看到参考8)
6。摘要:三种口味的E1cB
这三个例子遵循三种速率常数的三种可能性:k1(去质子化),其反向(k - 1),消除(k2)
实验区分这些不同的“味道”的E1cB从E2可以确定通过研究动力学同位素效应,使细微变化的身份离去基团。
还有的一种变体E1cB被称为E1cB离子对我没有这里讨论,讨论参考。
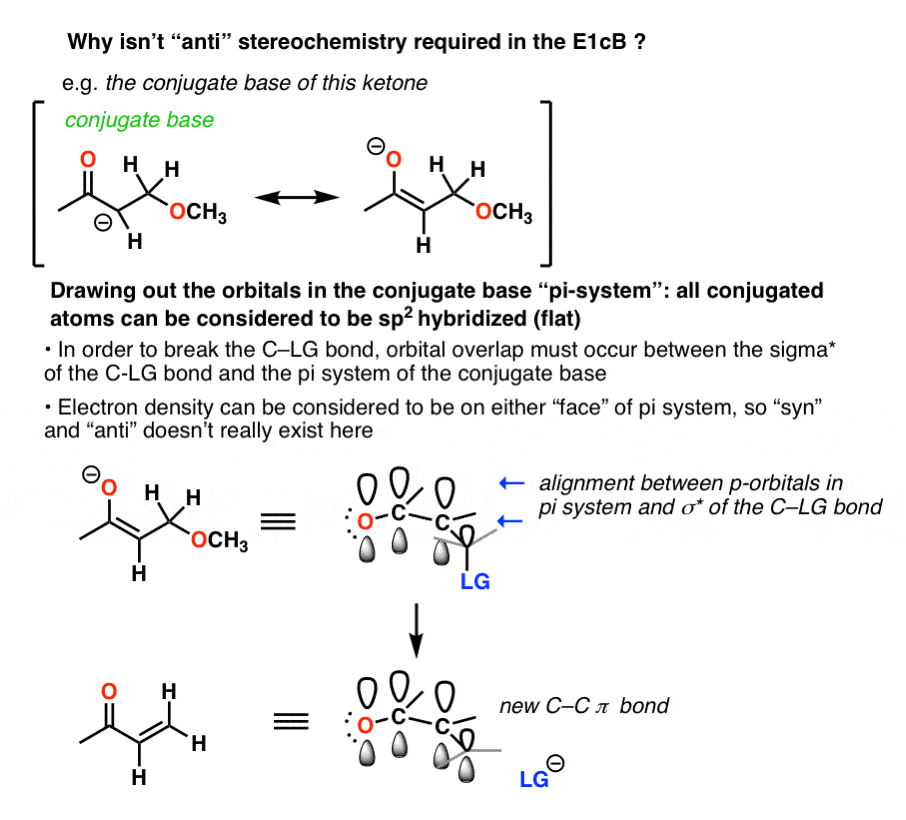
7所示。立体化学是什么?
的E2有一个严格的立体化学要求碳氢键和cx债券是反的。碳氢键必须反-periplanar C-LG债券,以便打破碳氢键的一对电子与C-LGσ*反键轨道。
通俗地说,就是你需要排队,碳氢键C-LG这样一对电子推出的离去基团(而形成新的π键)。
没有轨道对齐?没有去质子化。
在E1cB然而,“syn”和“反”可能发生(也适用于消除反应E1,如果你会记得)
的E1cB碳氢键时机制运作合理酸性。虽然基质可能四面体(sp3)开始,一旦共轭碱(离子)形成,它通常采用平面(三角形平面,sp2)结构,电子密度在两个“面孔”。的方向离去基团对阴离子并不重要,只要C-LG反键轨道与π体系保持一致。
这是因为一双打破碳氢键的电子捐赠的反键轨道cxσ*键。
在醛醇脱水的情况下得到一个阴离子。孤对p轨道。p轨道可以被认为是电子密度的分子,因为要么脸上有电子密度,cx债券是可以打破的。
(高级)引用和进一步阅读
理解“经典”之间的差异E1,E2,E1cB反应很简单。然而,了解这些机制之间的差异是决定冒险在核心物理有机化学。这些都是研究生论文!
一般的讨论E1cB,请参阅现代物理有机化学通过Anslyn和多尔蒂,第十章,3月份的先进的有机化学。
-
- 负碳离子机制烯烃消除得很深
d . j . McLennan
问:启化学。Soc。,1967年21岁,490 - 506
DOI:10.1039 / QR9672100490
一个广泛的概述E1cB机制有许多例子。 - β-Elimination 9-Fluorenylmethanol的水溶液:一个E1cB机制
r·a·O 'Ferrall,剃刀
j .化学。Soc。B。B1970年,260年
DOI:10.1039 / J29700000260
详细机械研究的经典E1cB反应,消除从9-fluroenylmethanol氢氧离子。 - 之间的关系E2和E1cBβ-Elimination机制
r·a·O 'Ferrall
j .化学。Soc。B。1970年,274年
DOI:10.1039 / J29700000274
第一篇论文“O 'Ferrall-Jencks阴谋”,一个有用的概念模型为研究之间的关系E1,E2,E1cB机制。 - Dihaloethylenes基本消除反应的动力学和机理反式消除
西德尼。米勒和理查德·m·诺伊斯
j。化学。Soc。1952年,629年
DOI:10.1021 / ja01123a015
本文作者确定消除各种速率常数独联体- - - - - -和反式- - - - - -dihaloethenes,发现消除的独联体dihaloethenes相当快。这里的结论是,消除的独联体-dihaloethenes通过协调一致的过程(E2),而消除的反式-dihaloethenes发生通过一个阴离子中间(记住:反式dihaloethenes, H和Clsyn彼此!) - 胺来自Dihalopropenes
a . t . Bottini b . j .国王和j·m·卢卡斯
j . Org。化学。1962年,27,3688 - 3690
DOI:10.1021 / jo01057a509
另一项研究中,作者显示炔烃可以由独联体和反式烯基陈词滥调。反应更容易当H和Br反式彼此(可以用氢氧化钠!),但需要NaNH2 / NH3当Br和H独联体。 比较流动反应的卤素dihalobenzenes氨酰胺钾
约瑟夫·f·Bunnett和弗朗西斯·j . Kearley Jr。
《有机化学》杂志上1971年36(1),184 - 186
DOI:10.1021 / jo00800a036
本文表明,形成芳炔的速率决定步骤取决于卤素的性质。去质子化病原的Br和我离开的离去基团F和Cl病原。这两个都是E1cB反应。
(本文也是独一无二的,据我所知,在自由诗体写)消除反应:实验确认的预测消除(.beta.-cyanoethyl)锍离子通过协调一致的,E2机制
Narinder Banait和威廉·p·Jencks
美国化学学会杂志》上1990年112年(19),6950 - 6958
DOI:10.1021 / ja00175a032
研究表明,与一个好的淘汰赛离去基团,E2之后,即使是一个强大的电子退出群。一阶base-initiated .beta. -消去反应涉及负碳离子中间
弗雷德里克·g·博德维尔、Kwok-Chun Yee和a·c·Knipe
美国化学学会杂志》上1970年92年(20),5945 - 5949
DOI:10.1021 / ja00723a022
的例子E1cB阴离子机制。
- 负碳离子机制烯烃消除得很深
我理解的醇醛缩合的负碳离子E1cB机制没有严格的立体化学的要求(因为负碳离子的电子参与共振与邻近的羰基)但你能通常假设sp2负碳离子?
如果没有邻近的电子撤回集团将负碳离子是sp3,暗示它需要一个反位置踢离去基?还是负碳离子(如二/三级胺)往往没有手性,因为他们可以自发地转化?或者做几乎所有E1cb机制需要周边电子撤回团体(如羰基或NO2)这意味着他们都有sp2性格像醇醛反应?
最后,你写的:“注意——很显然,当F或Cl离去基团,去质子化是缓慢的一步。然而,当离去基团Br,或者我去质子化是cx快的速率决定和破损。“我认为F或Cl你可能想说去质子化是一个“快”(而不是缓慢)的步骤。
谢谢!