烯醇和烯醇酯
烯醇酸催化的醛醇反应、卤化反应和Mannich反应
最后更新:2022年9月23日|
烯醇-醛醇缩合反应,卤化反应,Mannich反应
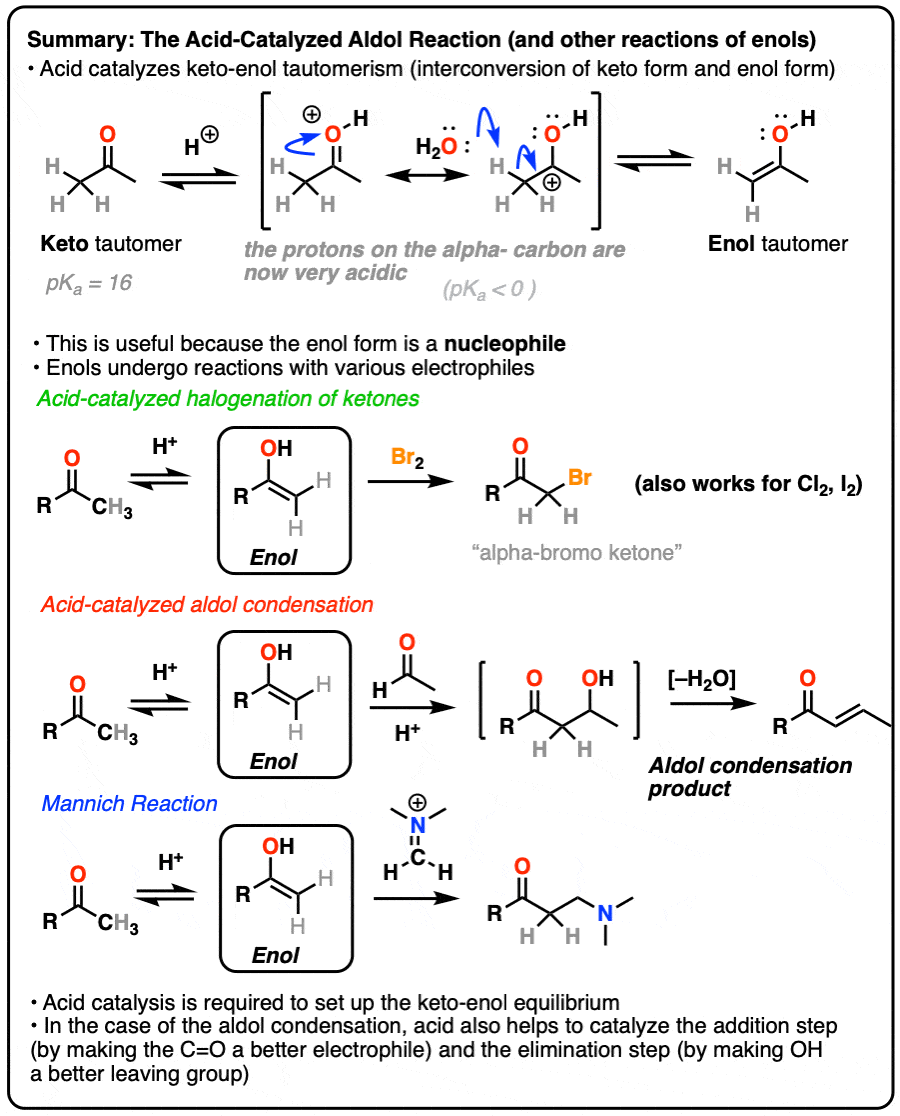
在之前关于keto-的文章中烯醇互变异构,我们讲过酮和烯醇形式。在这篇文章中,我们探讨的反应性烯醇互变异构体,可以作为亲核试剂在几个重要的反应中。
在这篇文章中,我们将有很多关于酸催化在帮助促进酮的作用烯醇互变异构,以及更多关于酸如何加速反应的讨论。的共轭酸是更好的亲电试剂"和"的共轭酸是更好的离去基团”。
目录
1.烯醇反应
在之前的文章中,我们探讨了酮烯醇互变现象(看到帖子:Keto-Enol互变现象,并描述了如何烯醇互变异构体亲核的另一个自我亲电酮互变异构体。在这篇文章中,我们将讨论一些更实际的方面,即:什么样的反应烯醇会发生反应吗?
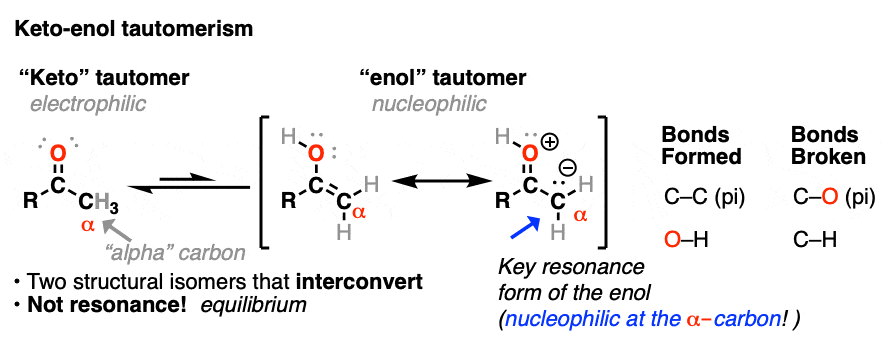
虽然亲核性不如烯醇酯[注1],烯醇确实在几个重要的有机反应中起着关键作用。
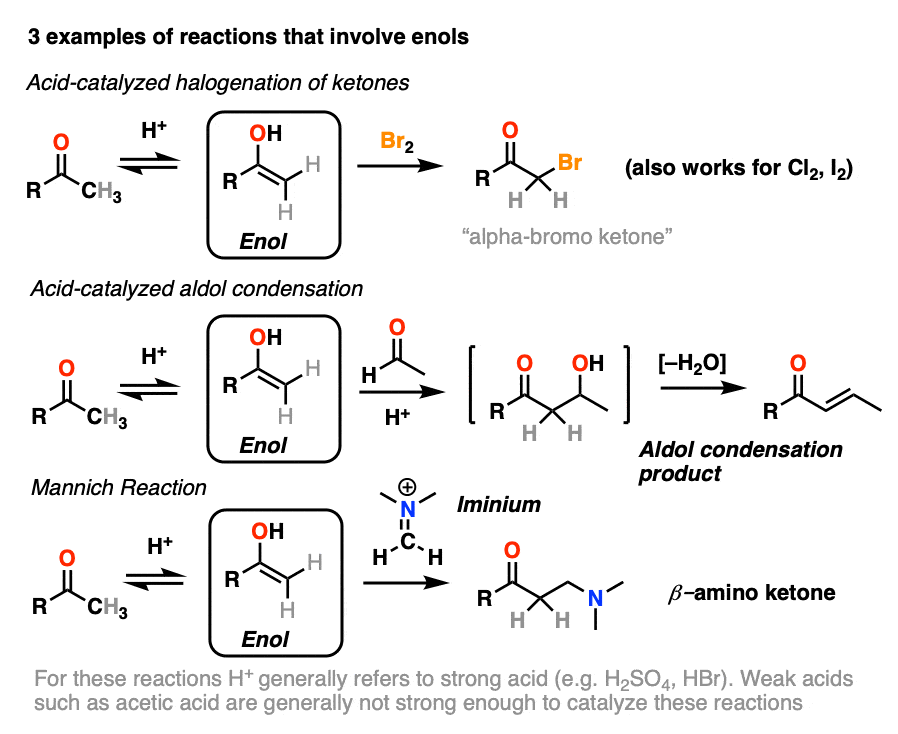
在这篇文章中,我们将看三个重要的例子:
- 酮的卤化
- 酸催化醛醇缩合。
- 曼尼希反应(它很像醛醇,除了烯醇加到碳氮键上而不是碳氧键上)
如果你仔细观察上面的三种反应,你会发现它们都是由酸催化.
为什么呢?让我们仔细看看。
2.碳的反应:聚二烷基化
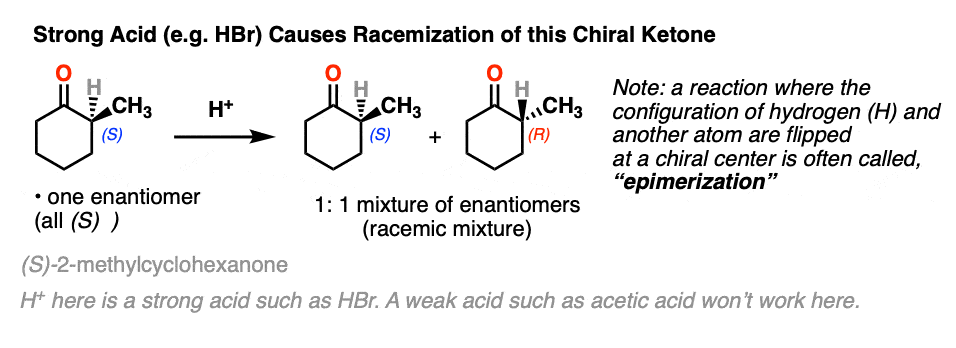
让我们以一个看起来并没有太多意义的反应为舞台。
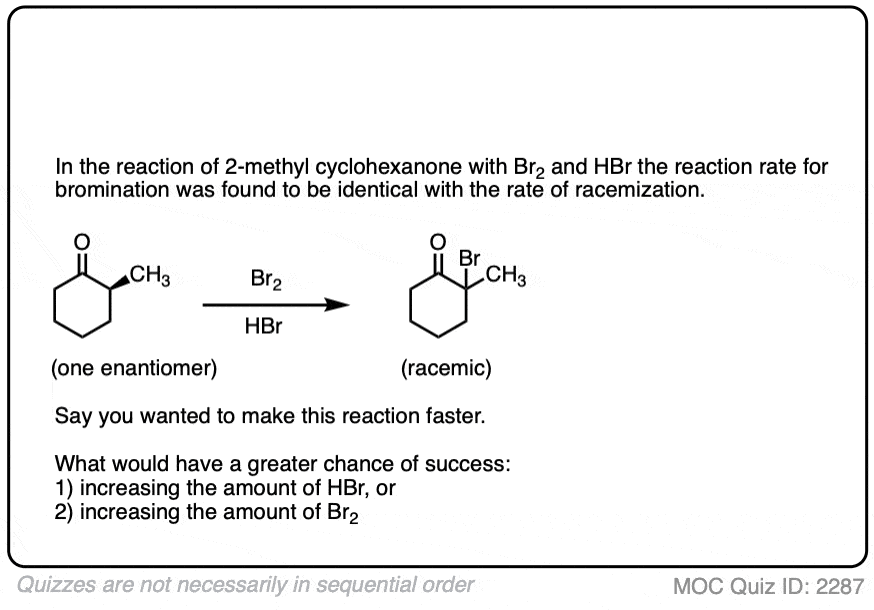
如果你用对映异构纯的溶液,具有光学活性酮比如这个[(年代)-2-甲基环己酮]加入几滴强酸,搅拌一夜,第二天早上你会发现它的光学活性已经变了完全消失了.[注2- - - - - -醋酸不够强,但几滴哈佛br就可以了]
它仍然是2-甲基环己酮,但是它变成了等量的(R)及(年代)对映体.一个外消旋混合物换句话说。(见文章:什么是外消旋混合物?)
相反,如果你取一个非常相似的对映体纯度酮(3-甲基环己酮)和执行完全相同的序列,光学活性将保持不变.
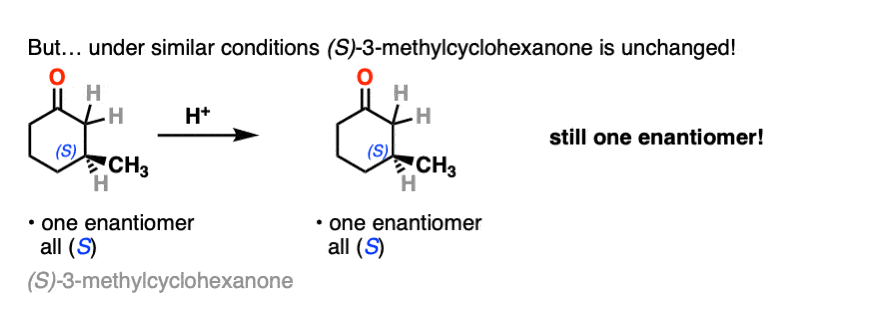
这是怎么回事?
两种酮都可以发生互变异构烯醇式中,C-2为三角平面(sp2杂化)。然而,烯醇2-甲基环己酮的形式,不再有手性中心。的烯醇形式是非手性的。
当这个平坦时,是非手性的烯醇然后被酸质子化再生2-甲基环己酮,它会这样做吗平等的可能性从顶部和底部的面,导致一个相等的(即外消旋)混合物(右)而且(年代)对映体.
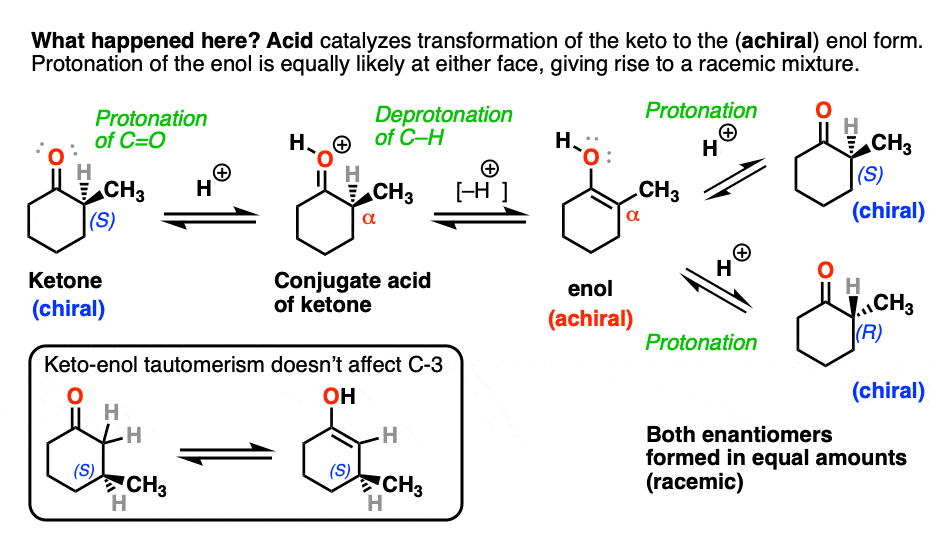
另一方面,……年代) 3-methylcyclohexanone保持其光学活性因为它的手性中心不会被破坏烯醇就形成了。
一个手性中心上的两个取代基互换(其中一个通常是H)的反应叫做差向异构化作用。
的外映异构化α碳酮和醛在酸和碱的存在下容易发生。碳链下游的碳不会受到影响,因为它只是α碳这是酸性的。
3.酮烯醇互变异构到底是如何被酸催化的?
同样的实验可以进行吗没有酸吗?是的,但它会慢得多。
酸有助于催化酮烯醇互变现象,允许快速平衡之间酮而且烯醇形式。
如何具体地说酸在这里有用吗?
如果你回到1号组织,你可能会发现一个深埋在你记忆库里的反应叫做E1的反应。(见文章:E1反应及其机理)
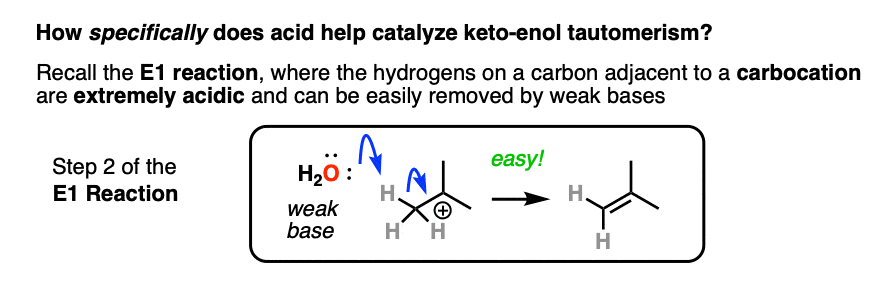
后离去基团叶子,下一步在E1是碳正离子旁边的碳的去质子化形成一个新的PI键。的E1需要这一步只需要一个弱碱(甚至水也可以!)自从离去基团已经离开了,碳有一个空的p轨道可以形成新的p键。(记住E2需要更强的碱才能得到它离去基团离开——参见文章:E2机制).
质子化的碳醛或酮非常类似于碳正离子.它有一个重要的共振形式形式电荷是在碳上,氧是中性的。[注3]
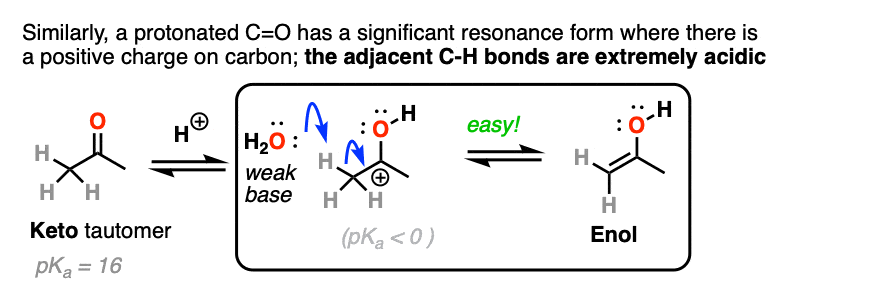
碳氢键的去质子化羰基(pK一个< 0)因此更容易而不是中性的情况醛/酮(pK一个= 16)
添加酸使C-H更容易脱质子化,从而形成烯醇容易得多。
酸也有助于转化烯醇回到酮的(缓慢)质子化反应α碳然后是O-H的(快速)去质子化。
用于酸催化转化烯醇酮,徘徊在这里然后会弹出一张图片或者点击这个链接.
{kind=link}
因为正向反应和反向反应都加速了,这就是我们说酸"帮助建立平衡"的意思。
现在我们来看一些烯醇的反应。
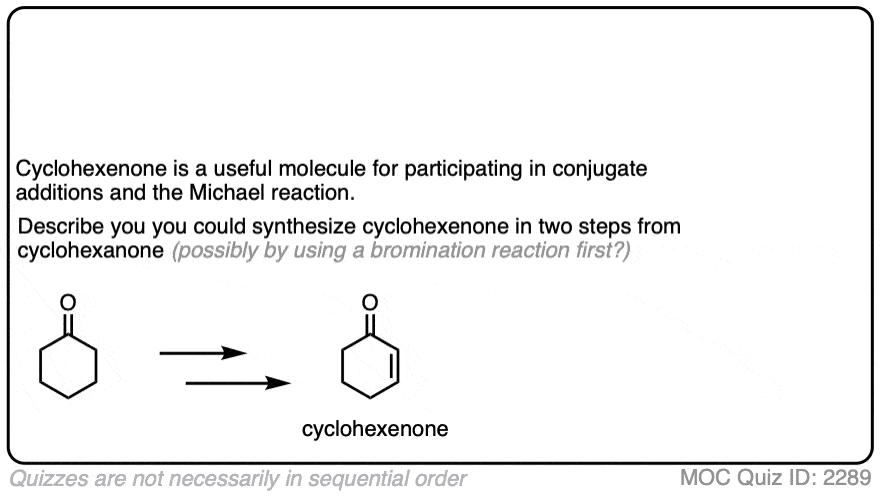
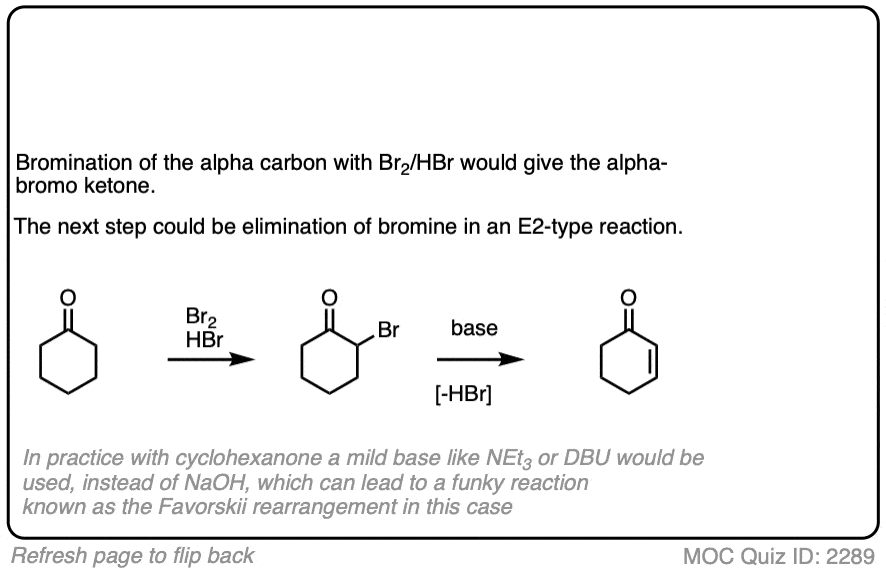
4.酮的溴化
烯醇实际上是富电子的烯烃,并会经历许多相同的反应。你们可能还记得烯烃会轻易地攻击卤素,如溴2例如,生成二卤化物。(看到帖子:烯烃的溴化)
如果你向a中加入强酸酮,并加入卤素(Cl2、溴2,我2),你会得到一个新的阿尔法光环酮.
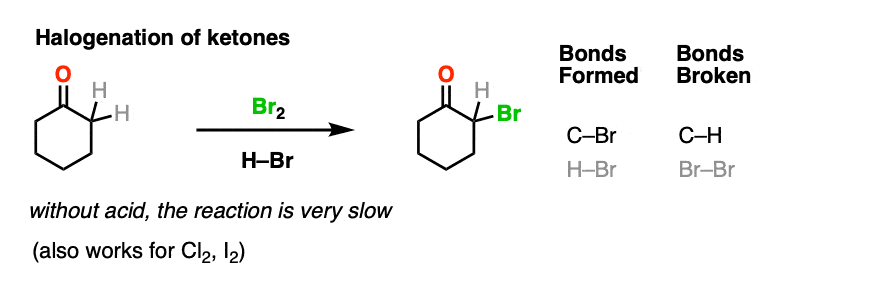
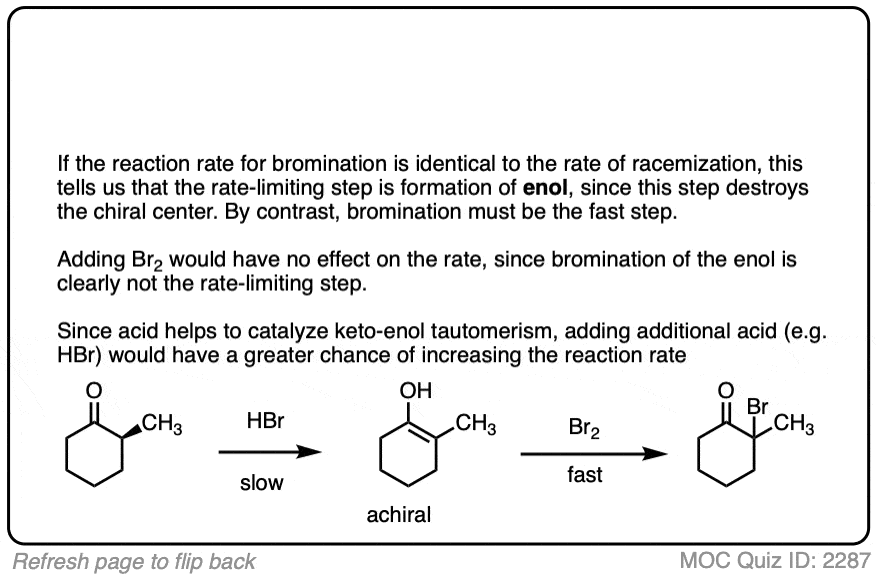
在没有酸的情况下,反应“极其缓慢”。[注意4此外,仔细的测量表明,添加过量的溴2不增加反应速率。
知道了这一点,一个有意义的机制是(缓慢的)转换酮对其烯醇形成,然后(快速)卤化。
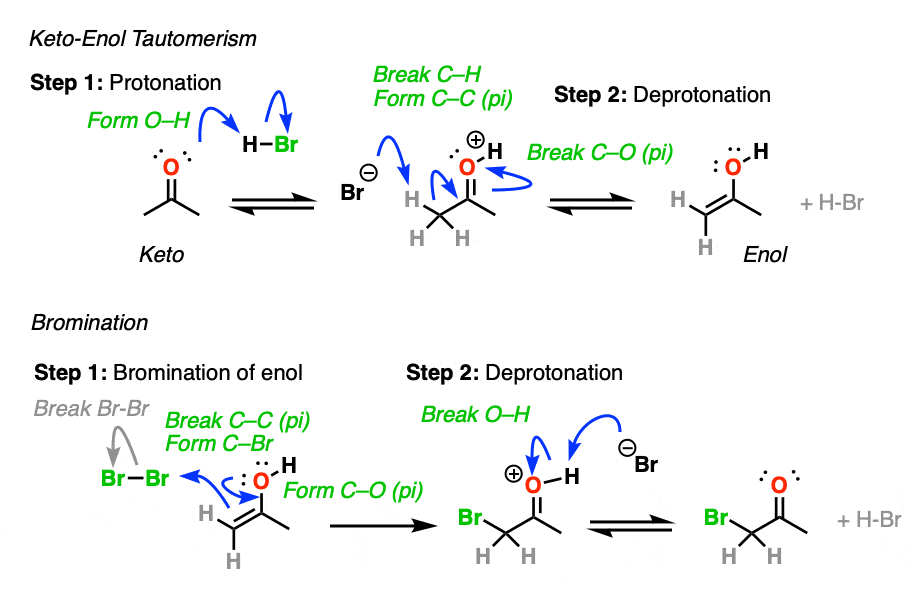
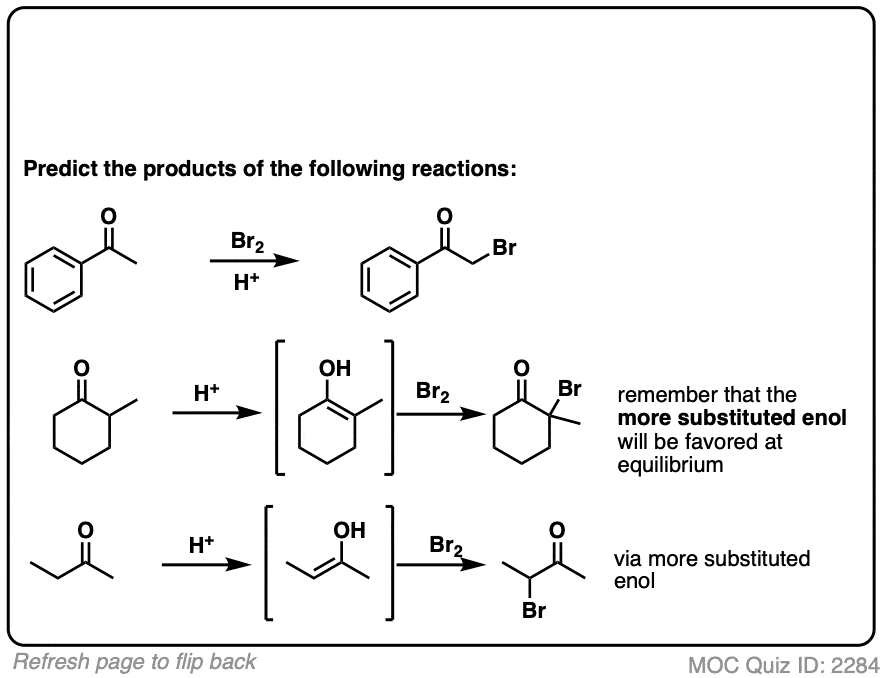
当不止一个烯醇能不能形成一样的酮,卤素会加入任意一个烯醇在平衡状态下,形式是最有利的(一般最替代-见以前的文章在酮烯醇更多细节为互变异构)。
 点击翻转
点击翻转
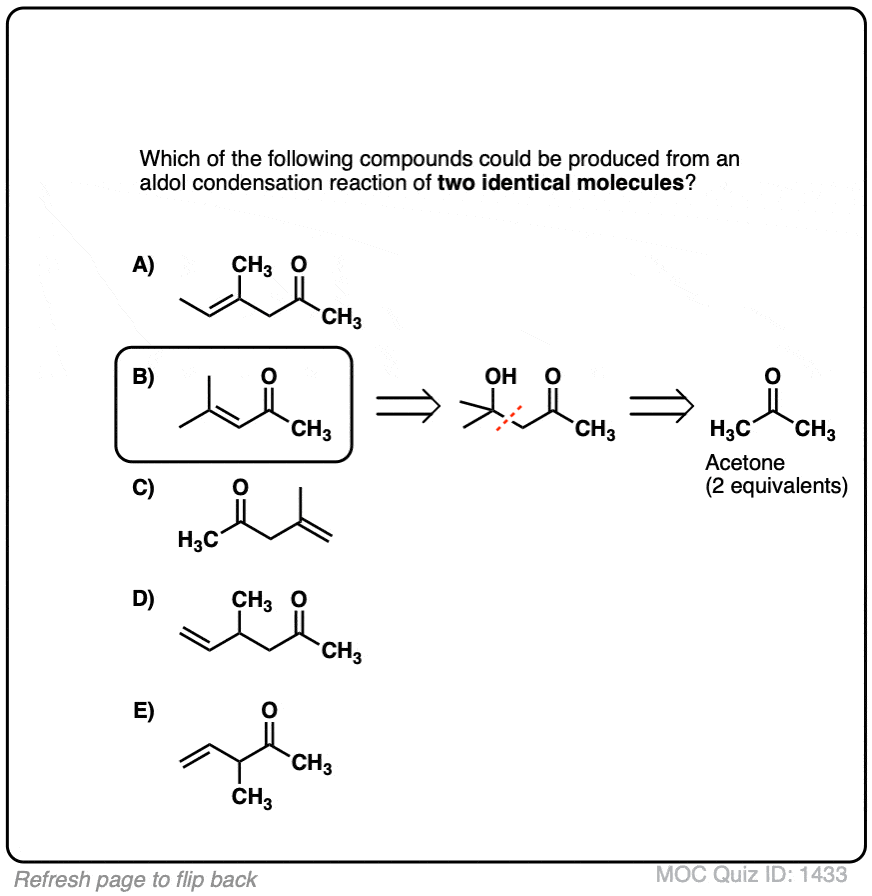
5.酸催化醛醇反应及其酸催化的三种途径
之前我们看到共轭碱烯醇(称为"烯醇化物)可以添加到醛类和酮类中,这种反应被称为醇醛反应.(见文章:醛醇加成和缩合)
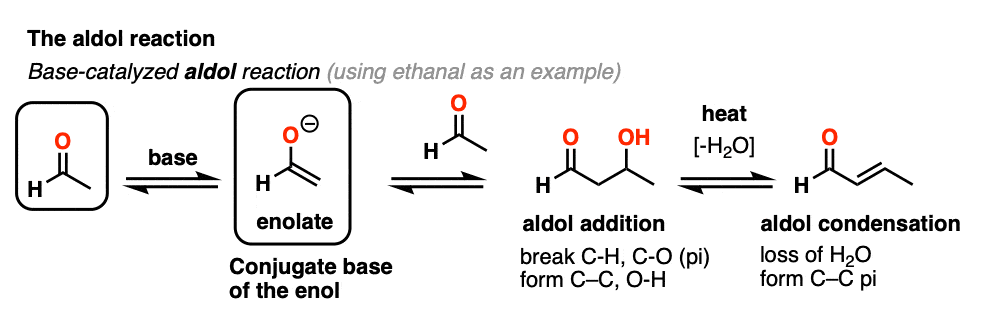
烯醇也可进行醛酮醛醇反应,如果酸性催化剂的存在有助于推动事情向前发展。酸催化的醛醇反应几乎总是导致冷凝产品(损失H2O),即使没有热量。
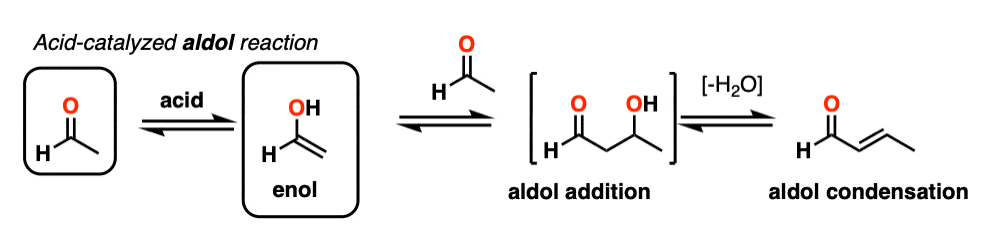
由于难以控制,酸催化的醛醇往往比碱催化的更少使用。[注5尽管如此,它还是很有教育意义的,因为它实际上突出了三个关键方法这种酸可以催化反应,这大致强调了这个反应的三个阶段。
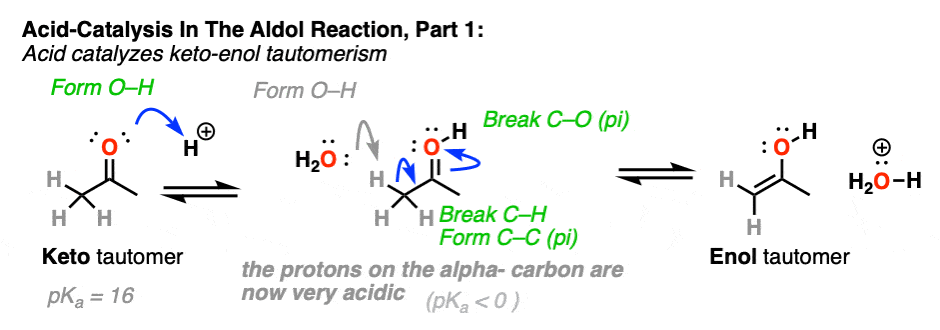
在第一阶段,酸催化酮烯醇互变异构现象,这为我们提供了我们的亲核试剂(烯醇),如前文所述。
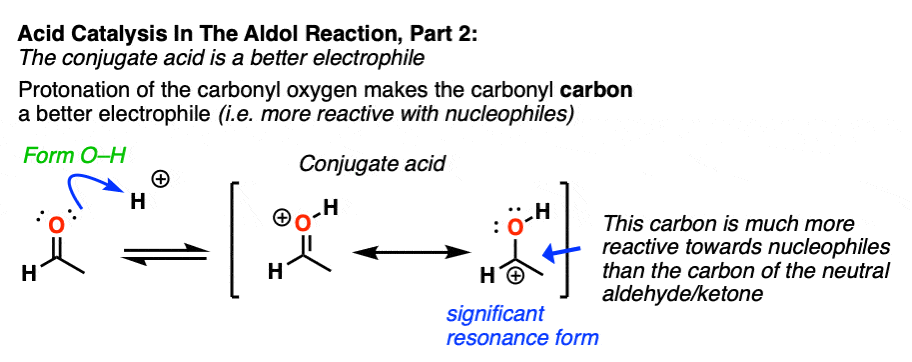
在第二阶段,酸使氧质子化醛或酮.由此产生的共轭酸的醛/酮是更好的亲电试剂在碳原子上,由于碳原子上带有正电荷的键共振形式的贡献[注3]。
的质子化了的醛/酮然后经过亲核试剂的加成烯醇,导致醇醛加成产物(脱质子后)。
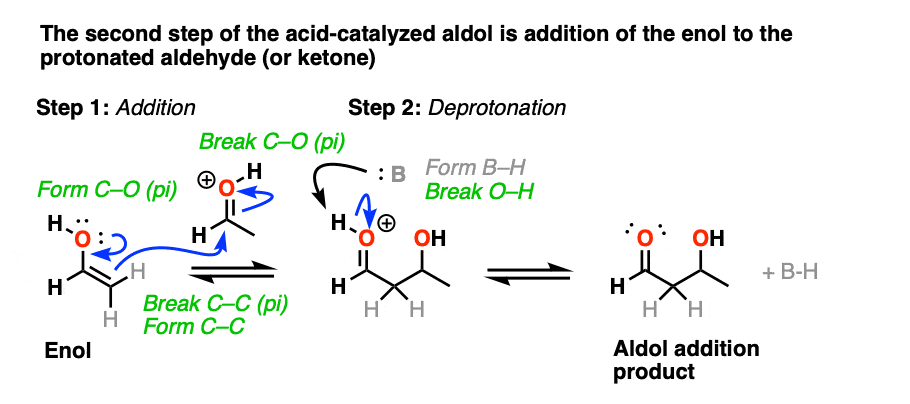
第三阶段的酸催化醛醇突出了如何共轭酸是更好的离开集团.加成产物羟基(OH)的质子化效果更好离去基团H2O。
在这一机制的一个版本中,水被“推出”烯醇加法的形式。同时形成C-O (pi)和迁移C-C (pi),可称为“1,4消除”或“共轭消除”步骤。还有其他方法可以说明这种情况,在讲义中有详细说明。[注6]
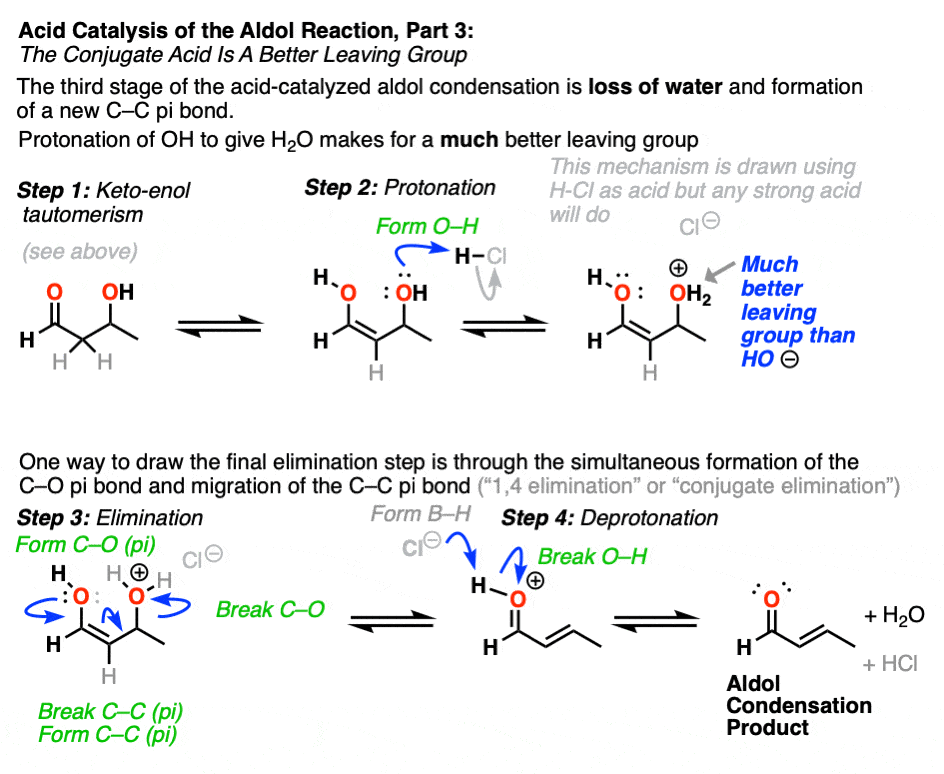
氧的去质子化产生中性醇醛缩合产品。
这里有几个羟醛缩合反应的例子,包括一个分子内酸催化羟醛缩合的例子。
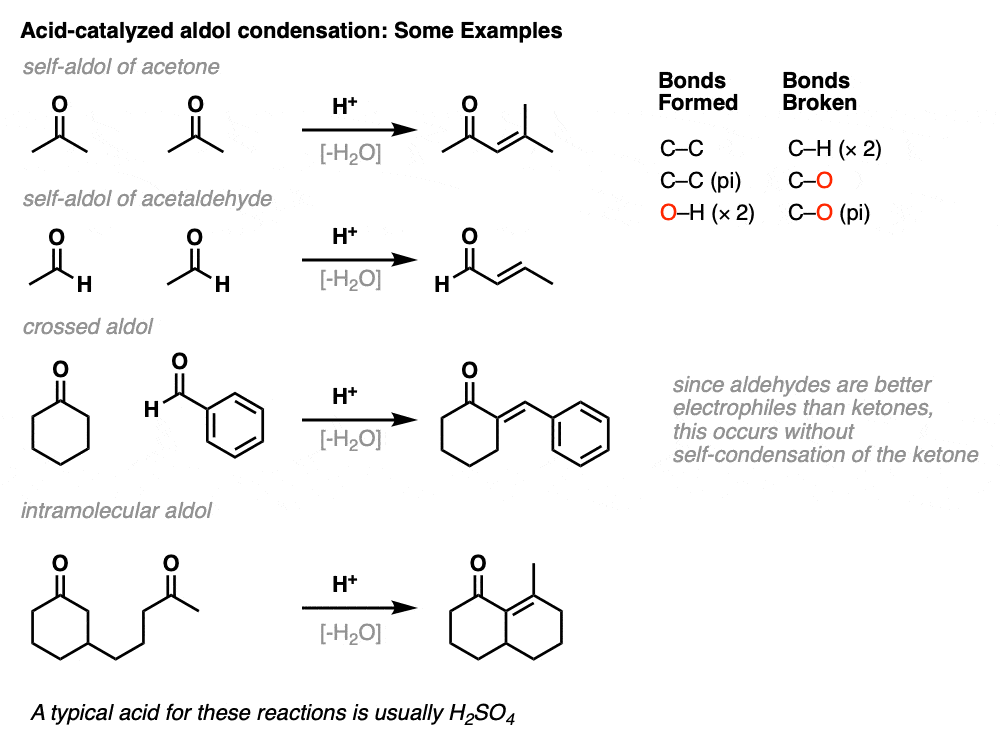
酸催化的醛醇反应也可以发生在分子内。例如,罗宾逊环结的一种变体使用了酸。(见文章:鲁滨逊环结)
6.曼尼希反应
曼尼希反应是醛醇反应的近亲,除了亲电试剂是C=N键而不是C=O键。
的亲电试剂在曼尼希中一般不是中性亚胺,而是带正电的iminium离子.
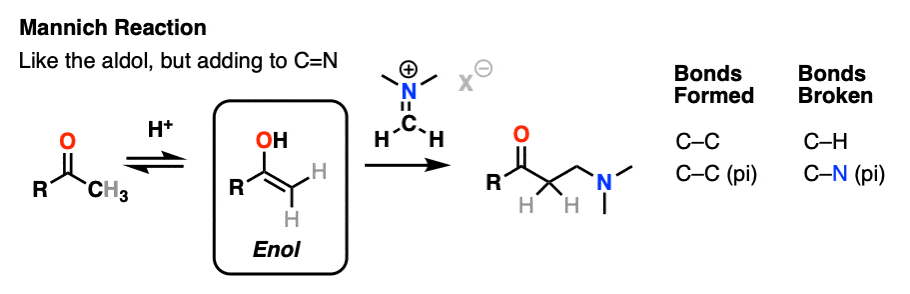
曼尼希反应的第一阶段是——你猜对了!-形成烯醇互变异构体。
在第二阶段,烯醇加到亚胺离子上。去质子化产生中性产物。
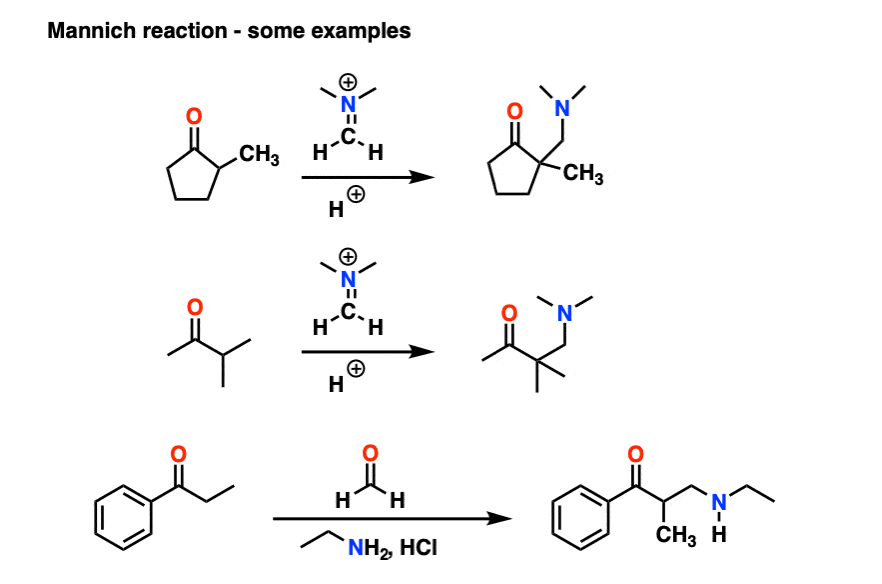
就像醛醇一样,如果在酸性条件下进行,反应会通过更多的取代体进行烯醇.
7.结论
那么我们学到了什么呢?一些事情。
- 烯醇关键的亲核中间体在吗卤化,酸催化醛醇反应,以及曼尼希反应.
- 酸催化酮烯醇互变异构现象;的α碳质子化的羰基比中性的酸性大得多醛或酮.
- 一个醛或酮手性中心在α碳将进行差向异构化作用在强酸的存在下。
- 对待一个醛或酮与Br2在强酸(HBr)的存在下会形成新的C-Br键α碳.在没有强酸的情况下不发生反应。
- 酸可以催化醇醛缩合.酸有助于促进酮烯醇互变异构,2)激活羰基面对攻击烯醇亲核试剂, 3)通过使生成的羟基变成更好的羟基,从而促进消除离去基团.
- 的曼尼希反应类似于醛醇反应,但涉及到一个烯醇到一个C = N而不是碳氧键。它也被酸催化。
笔记
注1- - - - - -烯醇不如烯醇酯亲核的共轭碱总是更好的亲核试剂而不是相应的中性物种。例如,RO(-)相对于ROH的一种量化方法是CH3.CH2O(-)的亲核性比CH高10个数量级3.CH2哦(见娃的网站这里是资料来源-你可能需要四处挖掘]。
没有类似的方法来比较亲核性烯醇类化合物和烯醇类化合物比较相似(它们比较难研究),但10个数量级可能是一个很好的猜测。
注2 -”醋酸不够强,但几滴哈佛br就可以了。”在一项研究中,发现对映异构物纯的外消旋化速度酮在醋酸/氯仿中非常缓慢,但在HBr存在的情况下非常快。(的酮是2-o-carboxybenzylindan-1-one,请参阅j .化学。Soc。1934773).
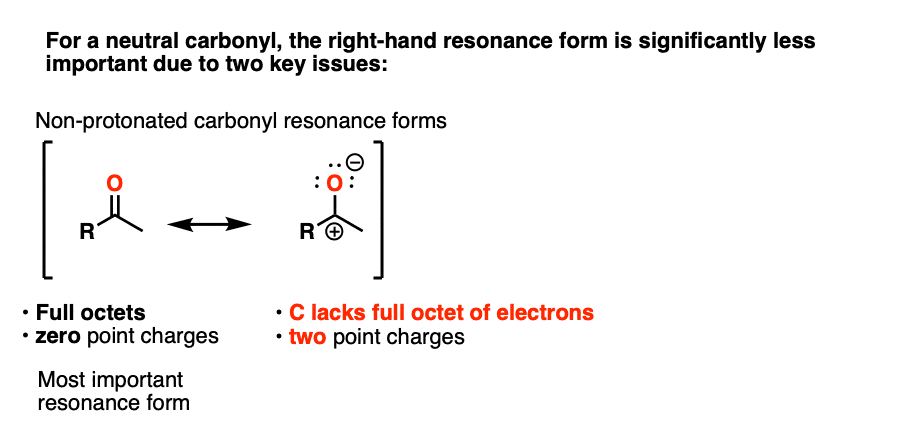
注3 -”它有一个重要的共振形式形式电荷是在碳上,氧是中性的"
一个直观的方法来衡量多少“碳正离子”字符酮比较了含碳正离子谐振形式对整体谐振杂化所作的相对贡献。(这里介绍了测量共振形式相对重要性的直观规则,4评价共振结构的规则年代)。这里有两个相关的关键项目是点电荷的数量(首选为零)和具有完整八位数的原子的数量(首选为零)。
在中性的情况下酮时,与碳正离子有共振形式两个点电荷(而C=O形式为零),此外C上缺少一个完整的八隅体。
一旦羰基质子化了的,这两个共振形式有相同数量点电荷(一个)。因此,碳缺乏一个完整的八隅体(碳正离子)的共振形式将是总体上没那么糟糕,因此,相对于非质子化,碳正离子对共振杂化的贡献更大羰基.
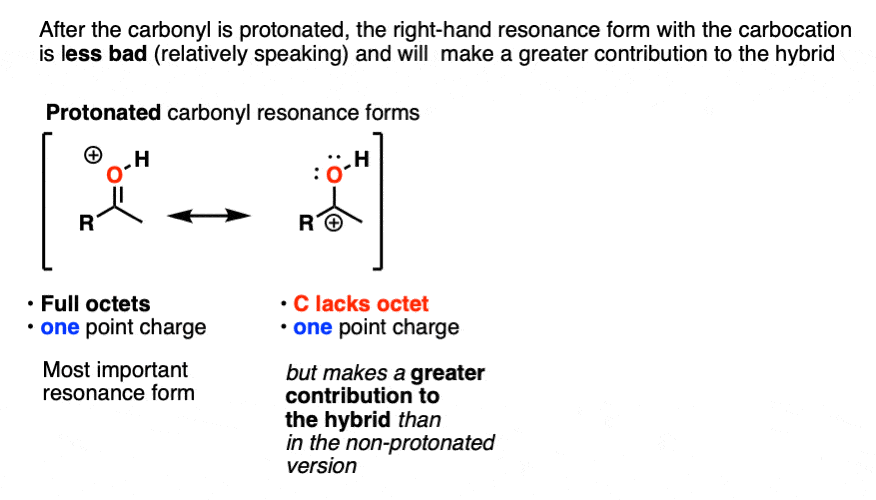
量化这种效应的一种方法是在红外光谱中检测C=O拉伸,因为键越强,波数越高。中性丙酮在1765厘米处有一个C=O拉伸-1而质子化丙酮的C=O长度为1580厘米-1.这表明C-O键明显减弱。
同样的论点也可以用来解释为什么带正电荷iminium离子是更好的亲电试剂而不是中性亚胺.
注4 -”在没有酸的情况下,溴化反应极其缓慢。”这是直接引用拉普沃斯(裁判)在他对酸催化溴化的开创性研究中。
注5 -”的酸催化的醛醇往往比碱催化的使用更少。”碱性促进醇得到更多的使用,因为它更容易控制烯醇化物可以通过使用适当的基底形成。在酸催化的醛醇中你被任何一种所控制烯醇形态在热力学上更稳定。
然而,还有一种醛醇反应叫做Mukaiyama醛醇反应它以一个烯醇醚随着亲核试剂,并使用强刘易斯酸(如TiCl4它在空气中会冒烟),以协助它们与醛的反应。
注6 -”还有其他方法可以说明这种情况,在讲义中有详细说明。”
通过共轭消除过程的消除是在乙醛的自缩合的研究中确定的(见裁判).
这一步在教科书中是如何显示的有一些变化,此外,在现实生活中也是如此。
其中一种表示方式是anE1-型反应,水离开,随后有一个去质子化事件。这可以发生;这是最正确的情况下,这将导致一个共振稳定的碳正离子(例如,毗邻一个富电子芳香环)。这里可以找到一个突出的例子。[裁判]
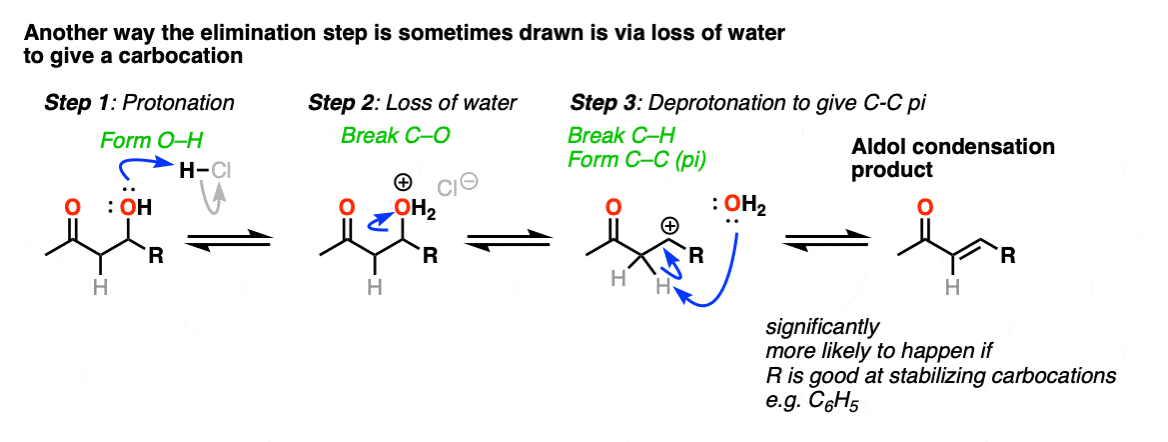
这远远超出了阅读这篇文章的人可能关心的范围,但也有OH基团发生取代的先例(可能是通过SN1)对好离去基团,随后用弱碱(或通过E1).以环己酮与盐酸的自醛醇缩合为例。
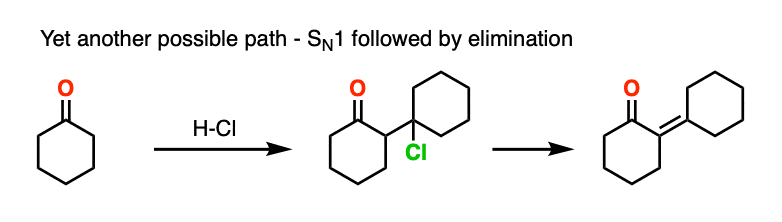
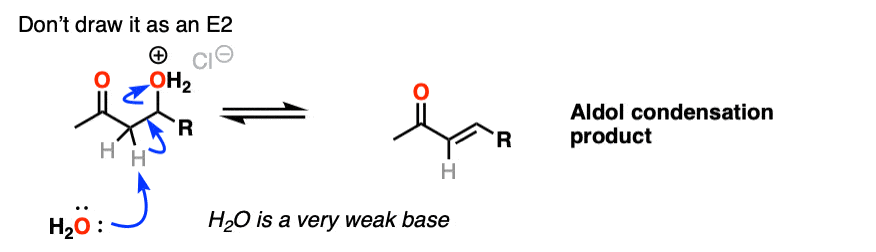
有一件事我可以放心地说,那就是你应该这么做不把它画成E2类型的反应。它需要C-H和C-OH2键的反平面排列,我不知道有任何证据表明这个过程依赖于立体化学(和你在图中看到的一样)E2).
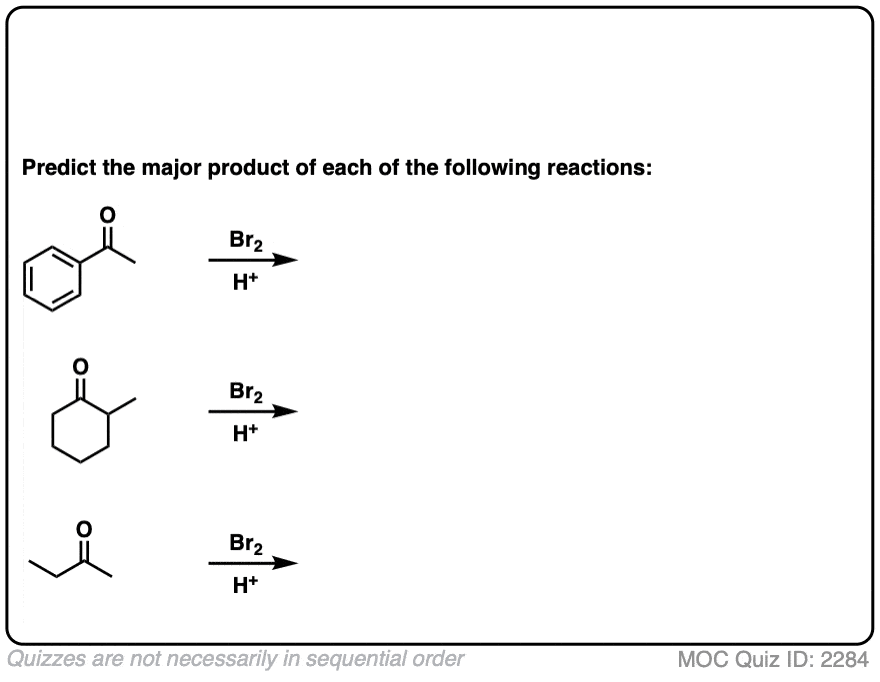
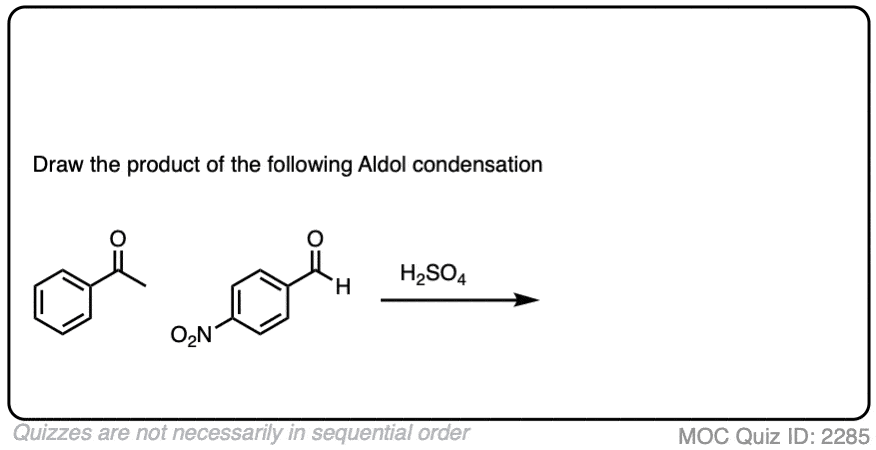
测试你自己!
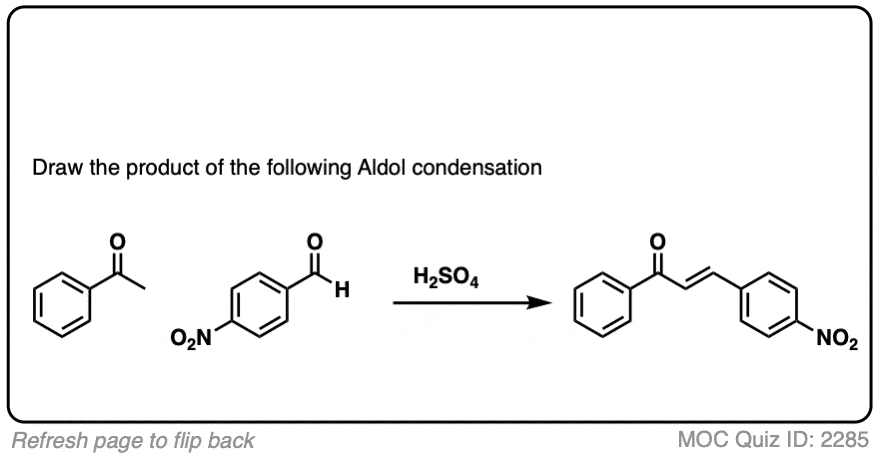
 点击翻转
点击翻转
 点击翻转
点击翻转

 点击翻转
点击翻转
 点击翻转
点击翻转
 点击翻转
点击翻转
 点击翻转
点击翻转
 点击翻转
点击翻转
(高级)参考资料和进一步阅读
《综合有机合成》第二卷对这篇文章很有用,马奇的《高级有机化学》、凯瑞和桑德伯格以及英戈尔德的《有机化学中的结构和机制》也很有用。
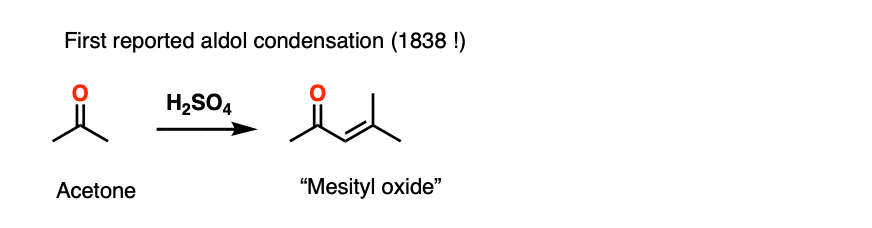
酸催化的醛醇反应自1838年以来就为人所知,这使它成为有史以来最古老的有机反应之一。第一个例子是二聚丙酮,从j . Prakt。化学1838,15,129.
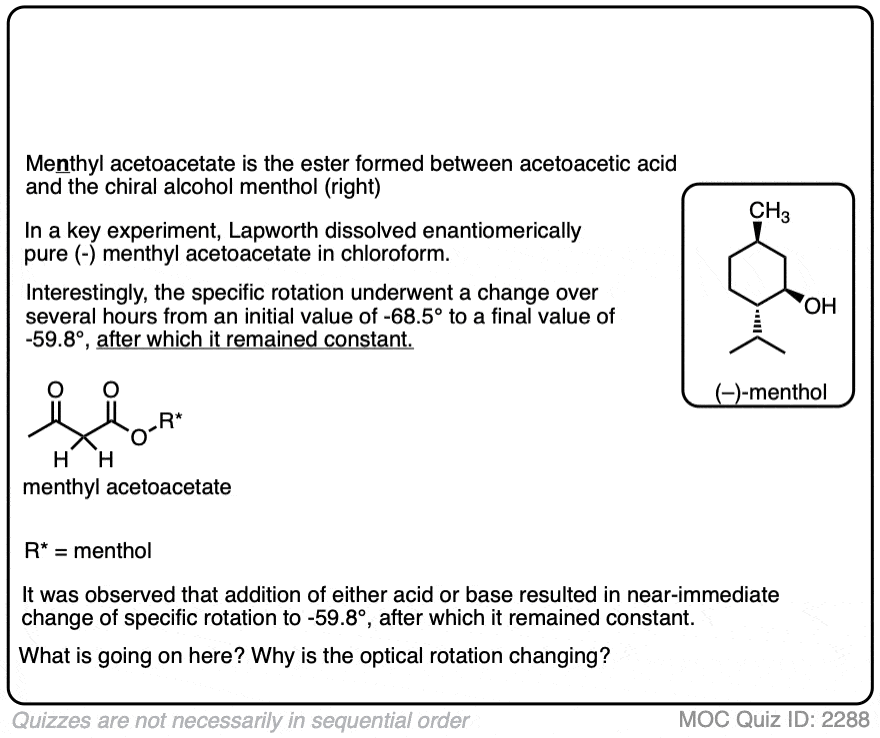
1.光学活性的酯类b -酮酸和b -醛酸第二部分。薄荷基乙酰乙酸盐
亚瑟·拉普沃斯和a·c·奥斯本·汉恩。
j .化学。Soc。,反式.1903,83, 1114 - 1129
DOI:10.1039 / CT9038301114
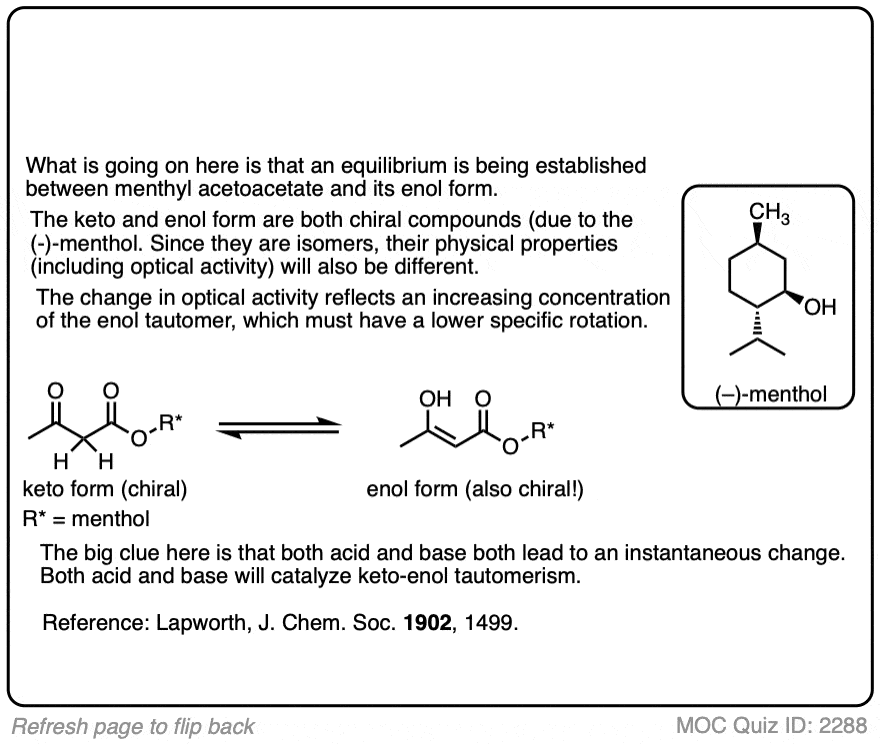
这是一篇非常有指导意义的论文,也是最早的例子之一,明确证明了酸在加速酮生成中的作用烯醇互变现象。作者准备了我n肌酸(不甲基)酯乙酰乙酸[(+)-薄荷醇是一种光学活性物质酒精很容易从自然来源获得],并测量了它的光学活性。他们发现我n肌酸酯经历了旋光改变在某种程度上,这取决于溶剂,突变速率高度依赖于酸。这证明了酮和酮之间平衡的建立烯醇形式。
2.卤素对含卤素化合物的作用羰基集团
亚瑟Lapworth
j .化学。Soc。反式.,190485年,30-42
DOI:10.1039 / CT9048500030
卤化的开创性研究羰基化合物。
“溴化丙酮在保持的条件下,最好将其视为在环境中发生的缓慢的、可逆的变化的结果丙酮由氢离子引起,然后是产物的溴化反应,这个变化是不可逆的。这个中间产物可能是烯醛形式的酮,因为它已经表明,在许多情况下,迅速达到平衡之间的互变异构形式羰基化合物是酸产生的。”
3.苯甲醛乙基甲基酮缩合反应(甲基乙酯缩合反应酮与苯甲醛)
C.哈里斯,G.汉斯·穆勒
的误码率。1902, 35(1), 966-971
DOI:10.1002 / cber.190203501156
甲基乙酯缩合酮在酸性条件下与苯甲醛缩合生成取代率较高的醛醇缩合产物!
4.与互变异构变化相关的光学活性。a的外消旋化速率和溴化速率的比较酮
c·k·英格尔德和c·l·威尔逊
j .化学。Soc。1934, 773年。
DOI:10.1039 / JR9340000773
溴化速率基本上与外消旋化速率相同,与之一致烯醇形成作为速率限制步骤。在氯仿和冰醋酸中,外消旋作用d-酮非常缓慢,但在溴化氢存在的情况下变得非常快”。
5.羰基反应。六、β-羟基酮脱水交替机制的证据1
唐纳德·诺伊斯和威尔默·l·里德
美国化学学会杂志195880(20), 5539 - 5542
DOI:10.1021 / ja01553a056
本文研究了酸催化甲基乙基醛醇反应酮还有各种取代苯醛。这里的关键是,最后的脱水步骤被建议通过烯醇当芳香环具有电子中性(Ph)或电子差(NO2)组在帕拉位置,但通过电离(即苄碳正离子形成)与富电子(OCH3)基团在帕拉的位置。陈述的原因是,从对甲基氧基到3-硝基的消除步骤的速率只有3倍的变化,这与人们所看到的不一致,如果它涉及a苄阳碳离子。
值得一提的是,唐纳德·诺伊斯的哥哥是物理学家罗伯特·诺伊斯他是英特尔公司的联合创始人。
6.羰基反应。8酸催化苯甲醛和对硝基苯甲醛与甲基乙基缩合反应动力学酮.ρ-σ相关性的一些观察1
唐纳德·s·诺伊斯和劳埃德·r·斯奈德
美国化学学会杂志195981(3), 620 - 624
DOI:10.1021 / ja01512a029
这是诺伊斯实验室1958-59年间一系列研究甲基乙基间酸催化醛醇反应的论文的第8部分酮取代苯醛。
7.乙醛的酸催化烯醇化和醛醇缩合
Lynn M. Baigrie, Robin A. Cox, Henryka slebokka - tilk, Michal Tencer和Thomas T. Tidwell
美国化学学会杂志1985107(12), 3640 - 3645
DOI:10.1021 / ja00298a039
广泛的物理有机化学研究了酸催化的简单醛醇缩合反应醛,乙醛。速率限制步骤是烯醇变成质子化乙醛。值得注意的是,他们的工作也与消除水发生从烯醇加法的形式。
另外两个有用的片段:“乙醛完全质子化到its共轭酸在H2所以4强于64%。“酮的烯醇化已知涉及两个水分子作为碱一起作用”。
8.酸催化罗宾逊退火
克莱顿·希斯科克,约翰·e·埃利斯,约翰·e·麦克默里,安东尼·科波利诺,
四面体信件,12 (52),1971, 4995 - 4996
DOI:10.1016 / s0040 - 4039(01) 97609 - 9。
本研究包含了几个酸催化罗宾逊环化的例子。
9.与2-甲基环戊酮和2-甲基环己酮的Mannich反应1
赫伯特·欧·豪斯和巴里·m·特罗斯特
有机化学杂志196429(6), 1339 - 1341
DOI:10.1021 / jo01029a016
几个有用的曼尼希反应的例子表明它们倾向于经过更多的取代烯醇.
1,4-消除机制对我来说是新的,似乎是一个好主意。但是,通常的1,2-消去机制并没有那么糟糕。当然,如果水不被用作溶剂,水作为基底是不好的[3月(第6版第1349页)说,即使在浓缩的H2SO4中,水也可以作为基底]。-OH2+是一个很好的离去基,H2O(或ROH,当使用醇时)接受质子形成一个氧鎓离子。这可以看作是E1的消除,而不是E2的消除。事实上,我在Ingold(第1004-1005页)中找到了关于这两种机制的讨论。有人认为Cl-离子作为碱代替水捕获质子形成HCl。但这在羟基溶剂中是不太可能的。我不知道哪种机制最有可能。不幸的是,今天没有化学家会做实验来阐明这一机制,因为这样的项目不会吸引资金。
如果它是一个直的E1,人们可能会看到重排,特别是在一个伯醇的情况下。由于从这些底物中消除水是容易的,首先发生烯醇化,然后从烯醇中给出一对电子的机制在我看来比E1型途径更合理,但根据取代模式,可能有不止一种机制可操作。例如,对于叔烯丙基醇,这可能是主要的反应机制。
感谢您花时间评论并添加来自March和Ingold的注释。
亲爱的先生,
我是研究员,研究方向是有机化学。本网站提供了关于有机反应和机理的极好信息。我给你留下了深刻的印象,非常感谢你的演讲。通过遵循这个网站,我得到预期的信息和知识。
感谢你,
你的真诚,
uttam帕蒂尔
谢谢你!
为什么醛醇缩合反应大多发生在碱的存在下
我认为碱是带走氢进行烯化所必需的。事实不是这样吗?因为在第二个途径中,你展示了引入酸会增加烯化速率。
是的,酮烯醇互变异构可以通过酸或碱的使用来辅助。
教授,在醛醇反应中哪一步是慢速决定的?
你能尽快回复吗??....急事……
谢谢
在酸催化醛醇加成反应中,缓慢的一步是烯醇对醛/酮的攻击。但如果你想了解更多信息,我建议:https://pubs.acs.org/doi/10.1021/ja01553a056
先生,为什么烯酸离子的氧不攻击羰基碳呢?我读过羰基碳是硬亲电试剂氧阴离子是硬亲核试剂,为什么这没有发生呢?
这在某种程度上可能会发生但产物是半缩醛,而半缩醛很容易还原到起始物质。所以通常都是死胡同。
在2-丁酮和2,2-二甲基丁烷的交叉醛醇反应中,考虑到2,2-二甲基丁烷缺乏酸性氢,2-丁酮可能是碳离子/亲核试剂。但从技术上讲,2-丁酮不是可以形成两种不同的亲核试剂吗,这不会导致两种不同的产物吗?我知道2-丁酮上的氢在3号碳上酸性更强,但是2-丁酮上1号碳上的氢是酸性的吗?它能不能变成亲核试剂,还是说它太不可能了以至于我们不考虑它?
尊敬的先生,我是一名研究学者,我的领域是有机合成,我用酸催化剂做醛酮反应,我用了2乙酰苯并呋喃和2,氨基,5,氯苯甲醛,但我期望的产品没有来,但产品来了2,氨基,5,氯苯乙酮,单晶xrd证实了结构,但它是如何形成的,在哪个机制,我想尝试找到机制请帮助任何建议,
我会再检查一下你的原料。
詹姆斯,
我非常喜欢这个帖子!谢谢你的工作。
在Clayden书2012 (pg 616)和Karty书(pg 912)中,酸催化脱水被称为E1,来自羰基,而不是来自烯醇。
我的感觉是这个机制涉及到一个E1,但是来自烯醇。事实上,在这种情况下,当水离开时,碳碳离子形成了三个共振贡献者的好处:一个带正电荷的碳,另一个带正电荷的羰基碳(双键转移了),加上氧碳离子。所以这个碳正离子有很多理由成为稳定的中间体,如果你正式地从氧碳离子中除去H+就得到了最终产物。
谢谢你的来信!
诺伊斯研究过:https://pubs.acs.org/doi/10.1021/ja01553a056
并得出结论,E1当然可以发生,特别是当一个相对稳定的碳正离子可以形成时(例如p-MeOC6CH4)。这种消除通常通过烯醇进行。乙醛的自缩合也是如此。看到的:https://pubs.acs.org/doi/10.1021/ja01553a056